一種金屬有機骨架納米復合材料的合成方法及其應用與流程
本發明屬于先進納米材料與納米技術領域,具體涉及一種金屬有機骨架(MOF)納米復合材料的合成方法及其應用,尤其涉及一種用于磷酸化肽富集以及糖肽富集與MALDI-TOF MS以及LC-MS/MS檢測的磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料的合成方法及其雙向應用。
背景技術:
蛋白質的糖基化以及磷酸化是生命過程中兩種重要且普遍的翻譯后修飾,它們與細胞間信號傳遞、細胞分裂、增殖、分化和相互作用等許多重要的復雜生物過程息息相關。一些研究表明,糖肽或磷酸化肽的表達水平異常,可作為很多疾病的生物標志,尤其是癌癥等人類重大疾病。所以對糖肽以及磷酸化肽的研究對疾病的早期診斷有著重要的意義。然而糖肽和磷酸化肽的豐度往往非常低,并且它們的質譜響應會受到高豐度非磷酸化肽/糖肽和蛋白質的壓制,樣品中的鹽分和表面活性劑同樣也會對其質譜行為產生干擾,使得其電離效率非常低,質譜檢測相對較為困難。因此在使用質譜方法分析復雜生物樣品中的糖肽和磷酸化肽段之前,對樣品中的肽段進行選擇性富集是十分必要的。
自19世紀80年代以來,一系列新的軟電離技術如快原子轟擊電離、基質輔助激光解吸電離、電噴霧電離等發現后,生物質譜技術迅速發展。由于質譜技術(Mass Spectrometry,MS)具有高準確性、高靈敏度和自動化操作的特點,并且它能夠準確測量肽段和蛋白質的相對分子量和氨基酸序列等,快速、精確地獲得多種蛋白質屬性參數,結合生物信息學工具,可迅速進行蛋白質的鑒定,從而為蛋白質的結構解析提供可靠依據。因此質譜技術無可爭議地成為當前蛋白質組學研究中不可或缺的平臺,質譜數據的信息質量直接決定了蛋白質鑒定的可靠性和鑒定數量。
隨著近年來研究的不斷深入,許多方法都被用來選擇性分離富集糖基化蛋白和多肽,比如硼酸親和色譜、肼化學、親水相互作用色譜(HILIC)、色譜分析法、體積排阻法等。其中HILIC方法應用最為廣泛,效果也較其他方法更好,HILIC方法中,例如金屬有機骨架材料、納米材料等等,都在糖肽富集方面引起了廣泛的重視。
此外,許多方法也都被用來選擇性分離富集磷酸化蛋白和多肽,比如免疫沉淀法、固相萃取法、超濾法、強陽離子交換色譜法、固定金屬離子親和層析法(IMAC)、金屬氧化物親和層析法(MOAC)等等。其中IMAC方法應用廣泛,效果也較好。通過金屬離子與磷酸化多肽上的磷酸基團間發生配位相互作用,從而起到富集磷酸化多肽的效果,MOF作為IMAC的一種,受到了廣泛的重視。
因此,本發明將HILIC與IMAC技術相結合,設計了一種磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料,通過鋯離子與磷酸化肽的緊密結合,以及親水相互作用與糖肽的緊密結合,達到一種材料雙向應用的效果。這種材料由于具有磁性,操作簡便、易于制得、且具備MOF的一管優異性能,在糖肽以及磷酸化肽富集方面有著很好的應用前景。
金屬-有機骨架材料是指由有機官能團為支架、金屬離子或金屬簇為中心節點,通過自組裝形成的具有規則納米孔道的三維周期性的網格結構多孔材料。具有非常大的比表面積、穩定的納米級孔道、可調控的孔道結構、良好的熱穩定性等優異性能,且不飽和的配位金屬可能與含有羧基、氨基等官能團的被分析物質發生配位作用,MOFs材料已成為有機化學、無機化學、物理化學領域的研究熱點。MOFs如今在氣體分離、苯及其同系物的選擇性吸附、藥物研發等發揮了其不小的作用。近年來,MOFs材料已初步應用于蛋白質/肽的分離富集,并顯示MOFs材料在蛋白質組研究中,如低豐度肽富集等有很好的潛力。
技術實現要素:
本發明的目的在于提供一種金屬有機骨架(MOF)納米復合材料的合成方法,尤其提出一種磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料的合成方法及其在糖肽以及磷酸化肽富集與MALDI-TOF MS以及LC-MS/MS檢測中的應用。
本發明提出一種金屬有機骨架(MOF)納米復合材料的合成方法,具體步驟如下:
(1)將FeCl3?6H2O溶于乙二醇中,磁力攪拌至澄清,加醋酸鈉充分攪拌,超聲后,轉移至反應釜中,在200℃條件下反應16小時后,冷卻,用去離子水及乙醇充分洗滌所生成的四氧化三鐵磁球,在50℃下真空干燥;
(2)將步驟(1)所得的四氧化三鐵磁球分散于三羥甲基氨基甲烷(Tris)緩沖液中,超聲15分鐘左右,加入多巴胺鹽酸鹽,在室溫下機械攪拌反應6-20小時,制得聚多巴胺包覆的磁球,用磁鐵分離產物后,用去離子水和無水乙醇充分洗滌,在50℃下真空干燥,得到聚多巴胺包覆的磁球;
(3)在二甲基甲酰胺中分散步驟(2)所得產物聚多巴胺包覆的磁球,超聲數分鐘,充分分散后加入氯化鋯,攪拌均勻,在反應溫度100-140℃下,反應15-45分鐘;
(4)在步驟(3)所得產物中加入配體2-氨基對苯二甲酸,在120℃下繼續加熱約15分鐘;
(5)步驟(4)所得產物用磁鐵分離后,用二甲基甲酰胺、蒸餾水和無水乙醇充分洗滌,在50℃下真空干燥,即得所需的金屬有機骨架(MOF)納米復合材料。
本發明中,步驟(1)中FeCl3?6H2O和乙二醇的配比范圍為(0.9-1.8)g:(50-100)mL。
本發明中,步驟(1)中FeCl3?6H2O和乙二醇的配比為1.35g:75mL。
本發明中,步驟(2)中Tris緩沖液的溶劑為去離子水和乙醇,體積比為1:1,pH值為8.5。
本發明中,步驟(2)中四氧化三鐵磁球和多巴胺鹽酸鹽的質量比為2.4:1。
本發明中,步驟(3)中聚多巴胺包覆的磁球和氯化鋯的質量比為(75-125):(120-200)。
本發明中,步驟(3)中聚多巴胺包覆的磁球和氯化鋯的質量比為5:8,反應溫度為120℃,反應時間為30分鐘。
本發明中,步驟(2)中所得產物聚多巴胺包覆的磁球和步驟(4)中的2-氨基對苯二甲酸的質量比范圍為(75-125):(90-150),反應溫度為100-140℃,反應時間為10-20分鐘。
本發明中,步驟(2)中所得產物聚多巴胺包覆的磁球和步驟(4)中的2-氨基對苯二甲酸的質量比為5:6,反應溫度為120℃,反應時間為15分鐘。
本發明還提出一種所述合成方法得到的金屬有機骨架(MOF)納米復合材料在糖肽富集與質譜鑒定中的應用,具體為:將金屬有機骨架(MOF)納米復合材料以超純水為溶劑配置成為10 mg/mL的材料分散液,將該材料分散液與目標糖肽溶液加入90%乙腈/1%三氟乙酸緩沖液中,混合并在酶解儀中孵育;通過離心分離出納米復合材料,用90%乙腈/1%三氟乙酸以及80%乙腈/1%磷酸緩沖液洗滌材料,隨后用30%乙腈/0.1%甲酸洗脫;取1μL洗脫液直接在MALDI-TOF MS進樣靶板上點靶,干燥后再點加1μL濃度為30mg/mL的2,5-二羥基苯甲酸(DHB)溶液于該液滴上,形成基質結晶,進行質譜分析。
本發明還提出一種所述合成方法得到的金屬有機骨架(MOF)納米復合材料在內源性磷酸化肽富集與質譜鑒定中的應用,具體為:將金屬有機骨架(MOF)納米復合材料配置成為10 mg/mL的材料分散液(溶劑為超純水),將該材料分散液與目標磷酸化肽段溶液加入50%乙腈/0.1%三氟乙酸緩沖液中,混合并在酶解儀中孵育;通過離心分離出納米復合材料,用50%乙腈/0.1%三氟乙酸緩沖液洗滌材料,隨后用0.4M氨水洗脫;取1μL洗脫液直接在MALDI-TOF MS進樣靶板上點靶,干燥后再點加1μL濃度為20mg/mL的2,5-二羥基苯甲酸(DHB)溶液于該液滴上,形成基質結晶,進行質譜分析。
本發明的有益效果在于:首次合成了結合傳統IMAC材料和HILIC(MOF)材料優點的納米復合材料,并成功應用于糖肽和磷酸化肽的分離富集。本發明提出的合成方法合成的磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料,合成方法簡單快速,鋯離子對磷酸化肽具有高靈敏度和高選擇性,氨基和糖肽可通過親水相互作用緊密結合,MOF提高了有效表面以及較好的體孔道結構。此納米復合材料可用于選擇性地富集生物樣品中的低豐度的糖肽和磷酸化肽,并用于MALDI-TOF MS以及LC-MS/MS檢測。由于MOF較高的表面積以及鋯離子與磷酸基團間的親和作用和氨基與糖肽間的親水相互作用,使得該納米復合材料可以對復雜生物樣品中的糖肽和磷酸化肽進行選擇性富集,大大提高了其質譜信號,本發明所提供的材料對磷酸化肽檢測限達20 amol/μL,糖肽檢測限達200 amol/μL,對非磷酸化肽段的選擇性達1:500(質量比),對非糖肽的選擇性達1:100(質量比),可直接從人體血清中檢測到4條內源性磷酸化肽,以及307條氮端糖肽對應于121種不同的糖蛋白。被富集肽段信噪比放大倍數高,具有較好的選擇性和高靈敏度,對復雜生物樣品中的糖肽和磷酸化肽的檢測有很好的。
附圖說明
圖1為實施例1制得的金屬有機骨架(MOF)納米復合材料的掃描電子顯微鏡照片以及透射電子顯微鏡照片;其中:SEM: a)為Fe3O4@PDA,b)為Fe3O4@PDA@UiO-66-NH2,TEM: c)為Fe3O4@PDA@UiO-66-NH2,d)為Fe3O4@PDA@UiO-66-NH2(放大圖);
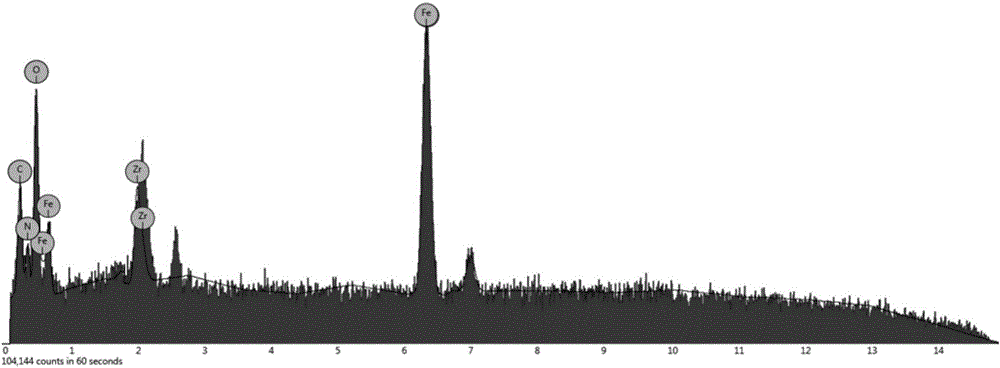
圖2為實施例1制得的金屬有機骨架(MOF)納米復合材料的能量色散X射線光譜及元素含量分布圖;
圖3為實施例1制得的金屬有機骨架(MOF)納米復合材料的氮吸附曲線;附圖:孔徑分布曲線;
圖4為實施例1制得的金屬有機骨架(MOF)納米復合材料的磁滯回曲線;
圖5為實施例1制得的金屬有機骨架(MOF)納米復合材料的親水性測試圖,其中(a)為5分鐘后,(b)為10分鐘后,(c)為30分鐘后,(d)為磁性分離3秒鐘后;
圖6為實施例1制得的金屬有機骨架(MOF)納米復合材料的紅外表征譜圖;
圖7為實施例1制得的金屬有機骨架(MOF)納米復合材料的拉曼表征譜圖;
圖8為實施例1制得的金屬有機骨架(MOF)納米復合材料的X射線衍射圖;
圖9為實施例1制得的金屬有機骨架(MOF)納米復合材料的Zeta電勢圖;其中:a) Fe3O4,b) Fe3O4@PDA,c) Fe3O4@PDA@UiO-66-NH2;
圖10為實施例2中經金屬有機骨架(MOF)納米復合材料富集前后的質譜圖,其中;a)是250 fmol/μL 的HRP酶解液富集前原液的質譜圖;b) 是250 fmol/μL 的HRP酶解液經金屬有機骨架(MOF)納米復合材料富集后洗脫液的質譜圖;c)1 pmol/μL 的IgG酶解液富集前原液的質譜圖;d)是1 pmol/μL 的IgG酶解液經金屬有機骨架(MOF)納米復合材料富集后洗脫液的質譜圖;e)是200 fmol/μL 的β-Casein酶解液富集前原液的質譜圖;f)是200 fmol/μL 的β-Casein酶解液經金屬有機骨架(MOF)納米復合材料富集后洗脫液的質譜圖;
圖11為實施例2中250 fmol/μL 的HRP酶解液經過金屬有機骨架(MOF)納米復合材料質譜圖,a)新鮮制得材料所得富集后洗脫液的質譜圖,b)材料循環使用5次后所得富集后洗脫液的質譜圖;200 fmol/μL 的β-Casein酶解液經過金屬有機骨架(MOF)納米復合材料,c)新鮮制得材料所得富集后洗脫液的質譜圖,d)材料循環使用5次后所得富集后洗脫液的質譜圖;
圖12為實施例2中250 fmol/μL 的HRP酶解液經過金屬有機骨架(MOF)納米復合材料質譜圖,a)新鮮制得材料所得富集后洗脫液的質譜圖,b)材料在-20℃保存1個月后所得富集后洗脫液的質譜圖;200 fmol/μL 的β-Casein酶解液經過金屬有機骨架(MOF)納米復合材料,c)新鮮制得材料所得富集后洗脫液的質譜圖,d)材料在-20℃保存1個月后所得富集后洗脫液的質譜圖;
圖13為實施例2中HRP酶解液經過金屬有機骨架(MOF)納米復合材料富集后質譜圖,HRP酶解液濃度為:a) 25 fmol/μL; b) 5 fmol/μL; c) 1 fmol/μL;d) 0.2 fmol/μL;β-Casein酶解液經過金屬有機骨架(MOF)納米復合材料富集后質譜圖,β-Casein酶解液濃度為:e) 25 fmol/μL; f) 1 fmol/μL;
圖14為實施例3中質量比為1:50的HRP和BSA酶解液的混合溶液a)富集前的質譜圖;b) 經過金屬有機骨架(MOF)納米復合材料富集后質譜圖;質量比為1:100的HRP和BSA酶解液的混合溶液c)富集前的質譜圖;d) 經過金屬有機骨架(MOF)納米復合材料富集后質譜圖;質量比為1:500的β-Casein和BSA酶解液的混合溶液e)富集前的質譜圖;f) 經過金屬有機骨架(MOF)納米復合材料富集后質譜圖;
圖15為實施例4中未經過酶解處理的健康人血清質譜圖,a)原液質譜圖,b)經過金屬有機骨架(MOF)納米復合材料富集后上清液質譜圖。
具體實施方式
下面的實施實例是對本發明的進一步說明,而不是限制本發明的范圍。
實施例1:一種磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料的合成。
(1)用乙二醇作為溶劑合成四氧化三鐵磁球,將1.35g FeCl3?6H2O溶于75mL乙二醇,磁力攪拌(手套封口)至澄清,后加3.6g壓碎醋酸鈉攪拌至溶解并繼續攪拌0.5h(手套封口)。超聲5min后,轉移至反應釜,200℃,16h。取出反應釜,冷卻過夜。倒出磁球,水洗5次(每次超聲5min)。用去離子水及乙醇充分洗滌磁球,到洗滌液清澈純凈,在50℃下真空干燥;
(2)配置三羥甲基氨基甲烷(Tris)緩沖液(溶劑為去離子水和乙醇,體積比1:1,pH=8.5),將步驟(1)所得的四氧化三鐵磁球120 mg分散于80mLTris緩沖液(內含0.05g Tris、40mL水、40mL乙醇)中,超聲15分鐘左右,加入0.32g多巴胺鹽酸鹽,室溫攪拌反應16h。磁鐵分離產物,用去離子水和無水乙醇充分洗滌,在50℃下真空干燥;
(3)在75 mL二甲基甲酰胺中分散步驟(2)所得產物100 mg,超聲一段時間,充分分散;加入氯化鋯160mg,攪拌均勻,120℃加熱攪拌30分鐘;
(4)在步驟(3)所得體系中加入配體2-氨基對苯二甲酸120mg,攪拌均勻,120℃加熱攪拌15分鐘,磁鐵分離產物,用二甲基甲酰胺、去離子水和無水乙醇充分洗滌,在50℃下真空干燥。
圖1為實施例1的掃描電子顯微鏡照片及透射電子顯微鏡照片。掃描電子顯微鏡圖可以看出在磁球外包覆了一層較薄的聚合物層,在修飾MOF后,表面的結晶形貌與聚合物的光滑層不同;透射電子顯微鏡圖可以看出聚合物@MOF層約為70納米厚;掃描電鏡型號為Philips XL30,將純化后的磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料均勻涂抹在導電膠上,噴金后進行SEM表征;透射電鏡型號為JEM-2100F(J0EL),將純化后的磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料的乙醇分散液滴在覆有碳膜的銅網上,干燥后進行透射電子顯微鏡觀察并拍照;
圖2為實施例1的元素分析,其中Zr元素的質量分數占8.0%,與預期一致,表如下;
圖3為實施例1的氮吸附曲線及孔徑分布曲線,由圖中可以看出,該納米復合材料具有較大的比表面積,且孔的大小為3.11納米左右;
圖4為實施例1的磁滯回線,雖然包覆了聚合物及MOF層之后,材料的磁響應有所下降,但仍保持著較高的磁響應強度,約為45.6emu·g-1;
圖5為實施例1的親水性測試圖,將材料分散在水溶液中形成穩定均一的水溶液,并在5分鐘、10分鐘、30分鐘后仍保持分散均一;而用磁鐵吸引后,則立刻變成澄清溶液與材料分離;
圖6為實施例1的紅外表征譜圖,該納米材料出現了較多的特征峰,如3400cm-1處的羧基特征峰、1500-1600cm-1處的苯環特征峰、560cm-1處的Fe-O-Fe振動峰;
圖7為實施例1的拉曼表征譜圖,<500cm-1處出現磁球的特征峰、1500-1600cm-1處在包覆多巴胺層后出現苯環特征峰,說明材料的成功合成;
圖8為實施例1的X射線衍射圖樣,2θ= 5.2, 7.0, 12.3, 18.2,22.3°是來自于MOF,而2θ= 30.3, 35.4, 43.2, 57.2,63.0°是來自于磁球內核,這也就說明磁性MOF材料的成功合成;X射線衍射儀型號為Bruker D4 X-ray diffractometer;
圖9為實施例1的Zeta電勢圖,Zeta電勢經過一層層包覆后先降后升是由于磁球表面被帶負電的鄰二酚羥基所占據導致電勢的下降,而之后MOF層的進一步修飾使表面帶正電導致電勢的上升,進一步說明材料表面的成功修飾。
實施例2:將實施例1得到的磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料作為固相微萃取吸附分離介質用于低濃度HRP酶解液以及β-Casein酶解液的富集與MALDI-TOF MS檢測。
(1)標準蛋白酶解液的制備:準確稱取2 mg HRP標準蛋白,用25 mM碳酸氫銨溶液配成濃度為 2 mg/mL的標準蛋白溶液,pH大約為8.3,煮沸十分鐘。按照質量比為1:50的胰蛋白酶與標準蛋白的比例,加入胰蛋白酶(trypsin),37°C孵育16小時,可得到2 mg/mL的HRP胰蛋白酶解液;準確稱取4 mg IgG標準蛋白,用25 mM碳酸氫銨溶液配成濃度為 4 mg/mL的標準蛋白溶液,pH大約為8.3,煮沸十分鐘。按照質量比為1:50的胰蛋白酶與標準蛋白的比例,加入胰蛋白酶(trypsin),37°C孵育16小時,可得到4 mg/mL的IgG胰蛋白酶解液;準確稱取2.5 mg β-Casein標準蛋白,用25 mM碳酸氫銨溶液配成濃度為 2.5mg/mL的標準蛋白溶液,pH大約為8.3,煮沸十分鐘。按照質量比為1:50的胰蛋白酶與標準蛋白的比例,加入胰蛋白酶(trypsin),37°C孵育16小時,可得到2.5 mg/mL的β-Casein胰蛋白酶解液。
(2)樣品的富集:
糖肽富集:用超純水配制10 mg/mL磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料的溶液。取20 μL的材料溶液于0.6 mL的離心管,用體積分數為90%乙腈和1%TFA的緩沖溶液洗滌2次后去除上清,加入用緩沖溶液稀釋后不同濃度的HRP酶解液(總體積為100μL),混勻,在37°C下震蕩富集30分鐘;離心分離材料,吸去上清液,用90%乙腈1%TFA溶液洗滌材料一遍,再用80%乙腈/1%磷酸溶液洗滌材料兩遍,然后加入10 μL的30%乙腈/0.1%甲酸溶液,37℃震蕩洗脫20分鐘,離心分離材料,吸出洗脫液備后用。
磷酸化肽富集:用超純水配制10 mg/mL磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料的溶液。取20 μL的材料溶液于0.6 mL的離心管,用體積分數為50%乙腈和0.1%TFA的緩沖溶液洗滌2次后去除上清,加入用緩沖溶液稀釋后不同濃度的β-Casein酶解液(總體積為100μL),混勻,在37°C下震蕩富集30分鐘;離心分離材料,吸去上清液,用50%乙腈0.1%TFA溶液洗滌材料三遍,然后加入10 μL的0.4 mol/L的氨水,37℃震蕩洗脫20分鐘,離心分離材料,吸出洗脫液備后用。
(3)點靶:取1 μL步驟(2)所述的洗脫液點到MALDI-TOF MS進樣靶板上,干燥后再點加1 μL濃度為30 mg/mL(糖肽)或20 mg/mL(磷酸化肽)的2,5-二羥基苯甲酸(DHB)溶液于該液滴上,形成基質結晶,干燥后再進行質譜分析。
(4)質譜分析以磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料作為固相微萃取吸附分離介質富集得到的糖肽和磷酸化肽并與富集前的原液質譜圖作對比。
濃度為250 fmol/ μL的HRP酶解液經過磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料富集后,質譜圖中出現了十九條歸屬于HRP的糖肽峰(m/z=1843.0, m/z=2541.4, m/z=2591.4, m/z=2611.4, m/z=3074.5, m/z=3087.7, m/z=3222.9, m/z=3321.8, m/z=3353.7, m/z=3369.7, m/z=3605.0, m/z=3672.1, m/z=3894.1, m/z=4056.2, m/z=4222.4, m/z=4719.6, m/z=4821.7, m/z=4838.7, m/z=4984.7)。
濃度為1 pmol/ μL的IgG酶解液經過磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料富集后,質譜圖中出現了二十一條歸屬于IgG的糖肽峰(m/z=2399.3,m/z=2431.3,m/z=2457.3,m/z=2488.3,m/z=2561.4,m/z=2602.4,m/z=2618.4,m/z=2634.4,m/z=2650.4,m/z=2764.5,m/z=2781.5,m/z=2796.5,m/z=2805.5,m/z=2837.5,m/z=2853.5,m/z=2926.6,m/z=2958.6,m/z=2967.6,m/z=3000.0,m/z=3130.0,m/z=3161.7)
濃度為200 fmol/ μL的β-Casein酶解液經過磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料富集后,質譜圖中出現了六條歸屬于β-Casein的磷酸化肽段的峰(m/z=1031.4,m/z=1279.1,m/z=1561.2, m/z=2061.9,m/z=2556.2,m/z=3122.5),四條去磷酸化峰(m/z=1963.9,m/z=2458.0,m/s=2927.3,m/z=3024.2)以及兩條來源于α-Casein的磷酸化肽段的峰(m/z=1466.7,m/z=1660.9,)。
實施例3:將實施例1得到的磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料作為固相微萃取吸附分離介質用于HRP酶解液或β-Casein酶解液和牛血清白蛋白(BSA)酶解液的混合溶液的富集與MALDI-TOF MS檢測。
(1)標準蛋白酶解液的制備:準確稱取2 mg 標準蛋白HRP、2.5 mg 標準蛋白β-Casein和5 mg標準蛋白BSA,用25 mM碳酸氫銨溶液配成濃度為2 mg/mL、2.5 mg/mL和5 mg/mL的標準蛋白溶液,pH大約為8.3,煮沸10分鐘。按照質量比為1:50的胰蛋白酶與標準蛋白的比例,加入胰蛋白酶(trypsin),37°C孵育16小時,可得到2 mg/mL的HRP胰蛋白酶解液、2.5 mg/mL的β-Casein酶解液和5 mg/mL的BSA酶解液。
(2)樣品的富集:
糖肽富集:用超純水配制10 mg/mL磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料的溶液。取20 μL的材料溶液于0.6 mL的離心管,用體積分數為90%乙腈和1%TFA的緩沖溶液洗滌2次后去除上清,先加入1 μL的2 mg/mL的HRP胰蛋白酶解液,分別按照HRP和BSA的質量比為1:50、1:100加入BSA酶解液,隨后加入相應體積的體積分數為90%乙腈/1%TFA的水溶液使體系配成總體積為100 μL的體系,混勻,在37°C下震蕩富集30分鐘;離心分離材料,吸去上清液,用90%乙腈1%TFA溶液洗滌材料一遍,再用80%乙腈/1%磷酸溶液洗滌材料兩遍,然后加入10 μL的30%乙腈/0.1%甲酸溶液,37℃震蕩洗脫20分鐘,離心分離材料,吸出洗脫液備后用。
磷酸化肽富集:用超純水配制10 mg/mL磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料的溶液。取20 μL的材料溶液于0.6 mL的離心管,用體積分數為50%乙腈和0.1%TFA的緩沖溶液洗滌2次后去除上清,先加入1 μL的2.5 mg/mL的β-Casein酶解液后,按照β-Casein和BSA的質量比為1:1:500加入BSA酶解液,隨后加入相應體積的體積分數為50%乙腈和0.1%TFA的水溶液使體系配成總體積為100 μL的體系,混勻,在37°C下震蕩富集30分鐘;離心分離材料,吸去上清液,用50%乙腈0.1%TFA溶液洗滌材料三遍,然后加入10 μL的0.4 mol/L的氨水,37℃震蕩洗脫20分鐘,離心分離材料,吸出洗脫液備后用。
(3)點靶:取1 μL步驟(2)所述的洗脫液點到MALDI-TOF MS進樣靶板上,干燥后再點加1 μL濃度為30 mg/mL(糖肽)或20 mg/mL(磷酸化肽)的2,5-二羥基苯甲酸(DHB)溶液于該液滴上,形成基質結晶,干燥后再進行質譜分析。
(4)質譜分析以磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料作為固相微萃取吸附分離介質富集得到的糖肽和磷酸化肽并與富集前的原液質譜圖作對比。
質量比為1:50的HRP和BSA的酶解混合液經過磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料富集后,從質譜圖中可以清楚地看到十二條來源于HRP的糖肽峰(m/z=2541.4, m/z=2591.4, m/z=2611.4, m/z=3222.9, m/z=3321.8, m/z=3353.7, m/z=3369.7, m/z=3672.1, m/z=4056.2, m/z=4222.4, m/z=4838.7, m/z=4984.7)
質量比為1:500的β-Casein和BSA的酶解液混合液經過磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料富集后,從質譜圖中可以清楚地看到五條來源于β-Casein的磷酸化肽段的峰(m/z=1031.4,m/z=1561.2,m/z=2061.9,m/z=2556.2,m/z=3122.5),四條去磷酸化峰(m/z=1952.0,m/z=2433.2,m/z=2927.6,m/z=3024.6)。
實施例4:將實施例1得到的磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料作為固相微萃取吸附分離介質用于健康人血清樣品中糖肽和磷酸化肽的富集與MALDI-TOF MS和LC-MS/MS檢測。
(1)樣品準備:
糖肽富集準備:2μL人體血清分散于198μL 25 mM碳酸氫銨溶液,煮沸10分鐘進行變形。后在60℃加入10 mM 二硫蘇糖醇(DTT)進行30分鐘還原反應,后在37℃在暗處加入20 mM 吲哚-3-乙酸(IAA)進行1小時烷基化反應。之后按照質量比為1:50的胰蛋白酶與蛋白濃度的比例,加入胰蛋白酶(trypsin),37°C孵育16小時,凍干待用。
磷酸化肽富集準備:用體積分數為50%乙腈和0.1%TFA的水溶液稀釋健康人血清樣品十倍。用體積分數為50%乙腈和0.1%TFA的水溶液配制10 mg/mL磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料的溶液。
(2)樣品的富集:
糖肽富集:用超純水配制10 mg/mL磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料的溶液。取40 μL的材料溶液于0.6 mL的離心管,用體積分數為90%乙腈和1%TFA的緩沖溶液洗滌2次后去除上清,加入100μL用90%乙腈和1%TFA的緩沖溶液稀釋的血清酶解凍干液,混勻,在37°C下震蕩富集30分鐘;離心分離材料,吸去上清液,用90%乙腈1%TFA溶液洗滌材料一遍,再用80%乙腈/1%磷酸溶液洗滌材料兩遍,然后加入10 μL的30%乙腈/0.1%甲酸溶液,37℃震蕩洗脫20分鐘,離心分離材料,吸出洗脫液凍干后備用。(LC-MS/MS)
磷酸化肽富集:用超純水配制10 mg/mL磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料的溶液。取20 μL的材料溶液于0.6 mL的離心管,用體積分數為50%乙腈和0.1%TFA的緩沖溶液洗滌2次后去除上清,在0.6 mL的離心管內加入10 μL的稀釋過的健康人血清,加入190 μL的體積分數為50%乙腈和0.1%TFA的水溶液,混勻,在37°C下震蕩富集30分鐘;離心分離材料,吸去上清液,用50%乙腈0.1%TFA溶液洗滌材料三遍,然后加入10 μL的0.4 mol/L的氨水,37℃震蕩洗脫20分鐘,離心分離材料,吸出洗脫液備后用。(MALDI-TOF MS)
(3)糖肽質譜分析:
LC-MSMS:由步驟(2)所得的凍干液分散在10 μL A相(H2O/0.1%FA)中。該儀器為EASY-nLC 1000 system并連有Orbitrap Fusion mass spectrometer。4 μL分散液根據線性梯度在110分鐘內從2% B相(乙腈/0.1%FA)到40% B相進樣入分析柱(C18, 75 μm x 50 cm)。色譜柱在最初狀態回穩10分鐘,柱流速為200 nL/min。激光電壓為 2.0 kV。Orbitrap質譜軟件在MS和MS/MS模式間自動切換。可達到m/z=200的分辨率。由質譜得到的數據基于2015年3月11日發布的Uniprot-SwissProt數據庫進行搜庫,碎片離子質量數容忍偏差度為0.050 Da,錯誤率(FDR)小于1%。
健康人血清經過磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料富集后,可辨識到307條氮端糖肽對應于121種不同的糖蛋白。
(4)磷酸化肽質譜分析:
點靶:取1 μL步驟(2)所述的洗脫液點到MALDI-TOF MS進樣靶板上,干燥后再點加1 μL濃度為20 mg/mL的2,5-二羥基苯甲酸(DHB)溶液于該液滴上,形成基質結晶,干燥后再進行質譜分析。
MALDI-TOF MS:質譜分析以磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料作為固相微萃取吸附分離介質富集得到的磷酸化肽并與富集前的原液和富集后的上清液的質譜圖作對比。
健康人血清富集前,由于受到嚴重的煩擾,無法看到內源性磷酸化肽的質譜峰,而經過磁球表面包覆聚多巴胺和氨基修飾的以鋯為中心金屬離子的金屬有機骨架(MOF)納米復合材料富集后,質譜圖中可以看到四條健康人血清中的內源性磷酸化肽的峰(m/z=1389.4,m/z=1460.5,m/z=1545.5,m/z=1616.5)。
- 還沒有人留言評論。精彩留言會獲得點贊!