氧吸留材料的制造方法及氧吸留材料與流程
本發明涉及氧吸留材料的制造方法及氧吸留材料。本發明特別涉及適合用在從汽車等的內燃機排出的廢氣的凈化用催化劑等中的氧吸留材料的制造方法及氧吸留材料。
背景技術:
在來自汽車等的內燃機的廢氣中包含氮氧化物(NOx)、一氧化碳(CO)、烴(HC)等,但這些物質可通過將CO及HC氧化、和將NOx還原的廢氣凈化催化劑來除去。作為該廢氣凈化催化劑的代表性的催化劑,已知的有使鉑(Pt)、銠(Rh)和/或鈀(Pd)等貴金屬載持于γ-氧化鋁等多孔金屬氧化物載體而成的三元催化劑等。
雖然可利用各種材料制造該金屬氧化物載體,但通常使用氧化鋁。但是近年來,為了利用載體的化學性質以促進廢氣的凈化,也提出了與氧化鋁組合或不與氧化鋁組合地使用氧化鈰(CeO2)、氧化鋯(ZrO2)和/或氧化鈦(TiO2)等各種其它材料。
例如,為了吸收廢氣中的氧濃度的變動從而提高三元催化劑的廢氣凈化能力,已在廢氣凈化催化劑中使用具有在廢氣中的氧濃度高時吸留氧、在廢氣中的氧濃度低時放出氧的氧吸留能力(OSC能力)的材料。作為具有氧吸留能力的材料,代表性的為氧化鈰。
為了利用三元催化劑的作用有效地進行CO及HC的氧化和NOx的還原,需要內燃機的空燃比為理論空燃比(化學計量比),因此為了發揮三元催化劑的廢氣凈化能力,優選利用具有氧吸留能力的材料來吸收廢氣中的氧濃度的變動并維持理論空燃比附近的氧濃度。進而,根據近年來的研究,發現氧化鈰不僅具有氧吸留能力,而且與載持于其上的貴金屬(特別是鉑)的親和性強,因此,可抑制該貴金屬的粒生長(燒結)。
這樣,關于廢氣凈化催化劑中的使用,氧化鈰具有優選的性質,但有時在其運用中不具有所需的耐熱性。因此,提出了將氧化鈰和氧化鋯固溶化,形成氧化鈰-氧化鋯系復合氧化物,從而提高耐熱性。
例如,在專利文獻1中,公開了一種氧吸留材料,其包含氧化鈰-氧化鋯系復合氧化物,在該氧化鈰-氧化鋯系復合氧化物中,通過鈰離子和鋯離子形成了燒綠石型的規則排列相(以下稱作“氧化鈰-氧化鋯系規則排列相”)。
氧化鈰-氧化鋯系規則排列相在還原狀態下為燒綠石相,在氧化狀態下為κ相。燒綠石相具有缺氧位點,氧原子進入該位點,由此燒綠石相變成κ相。另一方面,κ相通過放出氧原子而相變成燒綠石相。氧化鈰-氧化鋯系復合氧化物的氧吸留能力是基于在燒綠石相和κ相之間相互地相變而吸收和放出氧。
在使用氧化鈰-氧化鋯系復合氧化物作為廢氣催化劑的氧吸留材料的情況下,氧化鈰-氧化鋯系復合氧化物中所形成的氧化鈰-氧化鋯系規則排列相在濃(rich)狀態下變成燒綠石相,在貧(lean)狀態下變成κ相。通過在氧化鈰-氧化鋯系規則排列相的表面的氧的進出來產生這樣的相變。
如果氧化鈰-氧化鋯系規則排列相的粒徑粗大,則在氧化鈰-氧化鋯系規則排列相的內部,氧的進出是困難的。其結果,氧化鈰-氧化鋯系規則排列相的氧吸留速度下降,且氧吸留溫度上升。
現有技術文獻
專利文獻
專利文獻1:特開2009-84061號公報
技術實現要素:
發明所要解決的課題
根據專利文獻1,氧化鈰-氧化鋯系規則排列相的平均粒徑粗大至0.1~8μm。這是由于專利文獻1中公開的氧吸留材料的氧化鈰-氧化鋯系規則排列相是將氧化鈰和氧化鋯的復合氧化物粉末在1500℃以上的高溫下進行還原燒成而生成的。
本發明人發現了這樣的課題:將氧化鈰和氧化鋯的復合氧化物粉末在1500℃以上的高溫下進行還原燒成而得到的氧化鈰-氧化鋯系規則排列相由于其粒徑粗大而氧吸留能力不足。
本發明是為了解決上述課題而完成的,目的在于提供一種抑制氧化鈰-氧化鋯系規則排列相的粒徑變粗大的氧吸留材料的制造方法。另外,目的在于提供一種通過抑制氧化鈰-氧化鋯系規則排列相的粒徑變粗大來使氧吸留能力提高的氧吸留材料。
用于解決課題的手段
本發明人為實現上述目的,反復進行了專心研究,使得本發明得以完成。其主旨如下所述。
<1>氧吸留材料的制造方法,其包括:
在包含選自水溶性鑭鹽、水溶性鎂鹽和水溶性鈣鹽中的至少一種、水溶性鈰鹽、水溶性鋯鹽以及水溶性鋁鹽的水溶液中加入羥基羧酸,生成凝膠;
加熱上述凝膠,得到將上述鹽分解而成的固體生成物,
對上述固體生成物進行燒成,得到包含氧化鈰-氧化鋯系規則排列相前體和鋁酸鹽系復合氧化物前體的燒成物,
對上述燒成物進行還原熱處理,得到燒綠石相和鋁酸鹽系復合氧化物相互分散的第1復合體,和
對上述第1復合體進行氧化熱處理,得到使燒綠石相的至少一部分成為κ相的第2復合體。
<2><1>項中記載的氧吸留材料的制造方法,其中,上述固體生成物的燒成包括:
在使上述固體生成物中的有機物殘留的溫度下對上述固體生成物進行預燒成,得到預燒成物,和
在非活性氣體氣氛中對上述預燒成物進行再燒成,得到包含氧化鈰-氧化鋯系規則排列相前體、鋁酸鹽系復合氧化物前體和來自于上述有機物的碳化物的再燒成物,將該再燒成物供給上述還原熱處理。
<3><1>項中記載的氧吸留材料的制造方法,其中,上述還原熱處理包括:
將羥基羧酸和水加至上述燒成物,得到混合溶液,
使上述混合溶液干燥,得到干燥體,
在使上述干燥體中的有機物殘留的溫度下對上述干燥體進行準燒成,得到準燒成物,和
在非活性氣體氣氛中對上述準燒成物進行再燒成,得到包含上述氧化鈰-氧化鋯系規則排列相前體、上述鋁酸鹽系復合氧化物前體和來自于上述有機物的碳化物的燒成物。
<4><1>~<3>項的任一項中記載的氧吸留材料的制造方法,其中,在上述氧化熱處理之后,進一步進行再氧化熱處理。
<5><1>~<4>項的任一項中記載的氧吸留材料的制造方法,其中,上述鹽為硝酸鹽。
<6><1>~<5>項的任一項中記載的氧吸留材料的制造方法,其中,上述羥基羧酸為檸檬酸。
<7><1>~<6>項的任一項中記載的氧吸留材料的制造方法,其包括:
將選自釕、銠、鈀、鋨、銥、鉑、金、銅、鐵和鎳中的一種以上的金屬進一步載持于上述第2復合體。
<8>氧吸留材料,其中,至少一部分為κ相的氧化鈰-氧化鋯系規則排列相和鋁酸鹽系復合氧化物相互分散,
上述氧化鈰-氧化鋯系規則排列相通過X射線衍射測定的與(222)垂直的方向的平均粒徑為8.5~33nm。
<9><8>項中記載的氧吸留材料,其中,上述平均粒徑為18~25nm。
發明效果
根據本發明,能夠提供一種抑制氧化鈰-氧化鋯系規則排列相的粒徑變粗大的氧吸留材料的制造方法。另外,能夠提供一種通過抑制氧化鈰-氧化鋯系規則排列相的粒徑變粗大以使氧吸留能力提高的氧吸留材料。
附圖說明
圖1是示出通過本發明的制造方法得到的氧吸留材料的組織的示意圖。
圖2A是示出實施例1的氧吸留材料的X射線衍射譜的圖。
圖2B是示出實施例6的氧吸留材料的X射線衍射譜的圖。
圖2C是示出實施例7的氧吸留材料的X射線衍射譜的圖。
圖2D是示出實施例8的氧吸留材料的X射線衍射譜的圖。
圖2E是示出實施例9的氧吸留材料的X射線衍射譜的圖。
圖3是示出對實施例1和實施例6~實施例8以及比較例2~比較例3的氧吸留材料的氧化鈰-氧化鋯系規則排列相的(222)平均粒徑進行測定的結果的圖。
圖4是對于實施例1~實施例5和比較例1,示出由(鋁酸鹽系復合氧化物的摩爾數)/(氧化鈰-氧化鋯系規則排列相的摩爾數)表示的摩爾比與氧化鈰-氧化鋯系規則排列相的(222)平均粒徑的關系的圖。
圖5是對于實施例1和實施例6~實施例8以及比較例2和比較例3,示出I值={(111)峰的面積}/{(222)峰的面積}的圖。
圖6是示出在對實施例8的氧吸留材料進行透射電子顯微鏡觀察的結果(晶格像)中其第1視場的圖。
圖7A是示出由圖6的(a)所示區域的FFT(快速傅立葉變換;Fast Fourier Transform)譜和基于其的衍射模擬結果的圖。
圖7B是示出由圖6的(b)所示區域的FFT(快速傅立葉變換)譜和基于其的衍射模擬結果的圖。
圖7C是示出由圖6的(c)所示區域的FFT(快速傅立葉變換)譜和基于其的衍射模擬結果的圖。
圖8是示出在對實施例8的氧吸留材料進行電子顯微鏡觀察的結果(晶格像)中第2視場的圖。
圖9是示出由圖8的(a)所示區域的FFT(快速傅立葉變換)譜和基于其的衍射模擬結果的圖。
圖10A是示出對實施例9的氧吸留材料使用透射電子顯微鏡進行觀察的透射圖像的圖。
圖10B是示出對圖10A的觀察區域進行Ce-N EELS(EELS:Electron Energy Loss Spectroscopy;電子能量損失能譜)映射(mapping)的結果的圖。
圖10C是示出對圖10A的觀察區域進行Al-K EELS映射的結果的圖。
圖11A是示出實施例1和實施例10的氧吸留材料的每單位Ce質量%的H2消耗量分布的圖。
圖11B是示出比較例1和比較例4的氧吸留材料的每單位Ce質量%的H2消耗量分布的圖。
圖12是示出實施例1和實施例10以及比較例1和比較例4的氧吸留材料的每單位Ce質量%的H2消耗量的圖。
圖13是示出實施例9和比較例1的氧吸留材料的每單位Ce質量%的H2消耗量分布的圖。
圖14是示出實施例9和比較例1的氧吸留材料在各溫度下的每單位Ce質量%的H2消耗量的圖。
圖15是示出實施例1和實施例9的H2消耗量的圖。
圖16是示出對各氧吸留材料的室溫~950℃的H2消耗量各進行了3次測定時的各次的測定結果的圖。
圖17是示出實施例6和比較例5的氧吸留材料的每單位Ce質量%的H2消耗量分布的圖。
圖18是示出實施例15和比較例6的氧吸留材料的每單位Ce質量%的H2消耗量分布的圖。
圖19是示出實施例16和比較例7的廢氣凈化催化劑在400℃下的每單位Ce質量%的H2消耗量的圖。
附圖標記說明
1 氧化鈰-氧化鋯系規則排列相
1a κ相
1b 燒綠石相
2 鋁酸鹽系復合氧化物
具體實施方式
以下,詳細說明根據本發明的氧吸留材料的制造方法及氧吸留材料的實施方式。予以說明,以下示出的實施方式不限定本發明。
首先,對根據本發明的氧吸留材料的制造方法進行說明。
(凝膠的生成)
根據本發明的氧吸留材料的制造方法在包含選自水溶性鑭(La)鹽、水溶性鎂(Mg)鹽和水溶性鈣(Ca)鹽中的至少一種、水溶性鈰(Ce)鹽、水溶性鋯(Zr)鹽以及水溶性鋁(Al)鹽的水溶液中加入羥基羧酸,生成凝膠。
水溶性鈰鹽和水溶性鋯鹽為氧化鈰-氧化鋯系規則排列相(燒綠石相,κ相)的原材料。水溶性鑭鹽有時也成為氧化鈰-氧化鋯系規則排列相的原材料。予以說明,在該情況下,在氧化鈰-氧化鋯系規則排列相中,鈰的一部分被鑭置換。通過鑭的置換,即使在高溫氧化氣氛中,也能夠維持氧化鈰-氧化鋯系規則排列相。另外,鋯的一部分也可以被釔(Y)置換。通過釔的置換,在高溫氧化氣氛中,也能夠維持氧化鈰-氧化鋯系規則排列相。
水溶性鎂鹽和水溶性鋁鹽為鋁酸鹽系復合氧化物的原材料。水溶性鑭鹽有時也成為鋁酸鹽系復合氧化物的原材料。
關于這些原材料的配合,以在氧吸留材料中,氧化鈰-氧化鋯系規則排列相和鋁酸鹽系復合氧化物成為所期望的比例的方式將各自的原材料配合。
雖然在后面描述詳細內容,但在氧吸留材料中,通過氧化鈰-氧化鋯系規則排列相和鋁酸鹽系復合氧化物以納米水平相互分散,抑制了氧化鈰-氧化鋯系規則排列相進行粒生長。因此,作為原材料,在水溶性鈰鹽、水溶性鋯鹽和水溶性鋁鹽中配合選自水溶性鑭鹽、水溶性鎂鹽和水溶性鈣鹽中的至少一種,將這些鹽溶解在水中。
關于這些原材料的配合比,由(鋁酸鹽系復合氧化物的摩爾數)/(氧化鈰-氧化鋯系規則排列相的摩爾數)表示的摩爾比(以下有時簡單稱作“摩爾比”)同氧化鈰-氧化鋯系排列相的通過X射線衍射測定的與(222)垂直的方向的平均粒徑(以下稱作“(222)平均粒徑”)密切相關。如果以摩爾比為0.5以上的方式配合原材料,則(222)平均粒徑成為33nm以下,能夠明顯地確認粒生長的抑制作用。如果以摩爾比為1以上的方式配合原材料,則(222)平均粒徑成為25nm以下,能夠特別明顯地確認粒生長的抑制作用。另一方面,如果以摩爾比為3以下的方式配合原材料,則能在享有特別明顯的粒生長抑制作用的同時,在氧吸留材料中氧化鈰-氧化鋯系復合氧化物的相對量不過度減少。
作為水溶性鈰鹽,例如可舉出硝酸鈰、硝酸鈰銨、氯化鈰和硫酸鈰等。也可以使用它們的水合物。典型的為硝酸鈰及其水合物。
作為水溶性鋯鹽,例如可舉出硝酸氧鋯、氯氧化鋯、乙酸氧鋯等。也可以使用它們的水合物。典型的為硝酸氧鋯及其水合物。
作為水溶性鋁鹽,例如可舉出硝酸鋁、氯化鋁、硫酸鋁等。也可以使用它們的水合物。典型的為硝酸鋁及其水合物。
作為水溶性鑭鹽,例如可舉出硝酸鑭、硝酸鑭銨、氯化鑭和硫酸鑭等。也可以使用它們的水合物。典型的為硝酸鑭及其水合物。
作為水溶性鎂鹽,例如可舉出硝酸鎂、氯化鎂和硫酸鎂等。也可以使用它們的水合物。典型的為硝酸鎂及其水合物。
作為水溶性鈣鹽,例如可舉出硝酸鈣、硝酸銨鈣、氯化鈣和硫酸鈣以及鈣與鎂和/或鑭的鹽等。也可以使用它們的水合物。典型的為硝酸鈣及其水合物。予以說明,在水溶性鈣鹽中包含鑭的情況下,水溶性鈣鹽有時也成為氧化鈰-氧化鋯系規則排列相和鋁酸鹽系復合氧化物兩者的原材料。
除了目前為止說明的水溶性鹽以外,在不損害本發明的效果的范圍內,也可以追加另外的水溶性鹽。作為另外的水溶性鹽,例如可舉出水溶性釔(Y)鹽。水溶性釔鹽成為氧化鈰-氧化鋯系規則排列相的原材料。
作為水溶性釔鹽,例如可舉出硝酸釔、氯化釔和硫酸釔等。也可以使用它們的水合物。典型的為硝酸釔及其水合物。
予以說明,在以下的說明中,意味著也可以包含來自于在不損害本發明的效果的范圍內追加的水溶性鹽的元素。例如,在表示為“選自鑭、鎂和鈣中的至少一種、鈰、鋯和鋁”時,意味著也可以包含來自于在不損害本發明的效果的范圍內追加的水溶性鹽的元素。
在包含水溶性鑭鹽、水溶性鎂鹽和水溶性鈣鹽中的至少一種、水溶性鈰鹽、水溶性鋯鹽以及水溶性鋁鹽的水溶液中加入羥基羧酸,生成凝膠。作為羥基羧酸,只要能夠生成凝膠就不特別限定,但例如可舉出檸檬酸、酒石酸和乳酸等。典型的為檸檬酸。
(凝膠的加熱)
加熱得到的凝膠,得到將上述鹽分解而成的固體生成物。該固體生成物含有包含選自鑭、鎂和鈣中的至少一種的化合物以及包含鈰、鋯和鋁的化合物。這些化合物例如可以為氧化物和/或氫氧化物。
加熱溫度可根據鹽的種類來適當決定。在鹽為硝酸鹽的情況下,關于加熱溫度,優選為150~250℃。如果加熱溫度為150℃以上,則能夠分解硝酸鹽。更優選為165℃以上。另一方面,如果加熱溫度為250℃以下,則不會同時引起硝酸鹽的分解及其燒成。其結果,不會生成粗大的燒成物。更優選為200℃以下。關于硫酸鹽也可以同樣。
加熱時間可根據凝膠的量來適當決定。例如,加熱時間可以為10分鐘以上、20分鐘以上或25分鐘以上,可以為5小時以下、1.5小時以下或45分鐘以下。
予以說明,在用于將鹽分解的加熱后,為了使殘留的固體生成物干燥,也可根據需要將殘留的固體生成物放入電爐等中,在150~250℃下持續加熱20~30分鐘。
(固體生成物的燒成)
在此,關于固體生成物的燒成,雖然示出了實施方式A和實施方式B,但不受它們所限定。
<實施方式A>
在實施方式A中,通過對將鹽分解而成的固體生成物進行燒成,從固體生成物得到包含氧化鈰-氧化鋯系規則排列相前體和鋁酸鹽系復合氧化物前體的燒成物。燒成物中的前體以納米水平相互分散。
氧化鈰-氧化鋯系規則排列相前體是具有鈰、鋯和氧不規則排列的結構的氧化物。鋁酸鹽系復合氧化物前體是具有在成為完全的鋁酸鹽系復合氧化物之前的不完全結構的氧化物。
燒成溫度只要是能夠形成前體的溫度就不特別限定。關于燒成溫度,優選為400~850℃。如果燒成溫度為400℃以上,則能夠形成前體。更優選為600℃以上。進一步優選為700℃以上。另一方面,如果燒成溫度為850℃以下,則生成的前體不易粒生長。更優選為825℃以下。進一步優選為800℃以下。
燒成氣氛只要是能夠形成前體的氣氛就不特別限定,例如可舉出大氣。
燒成時間可根據固體生成物的量來適當決定。燒成時間可以為1小時以上、2小時以上或2.5小時以上,可以為10小時以下、6小時以下或3.5小時以下。
<實施方式B>
在實施方式B中,對將鹽分解而成的固體生成物在使該固體生成物中的有機物殘留的溫度下進行預燒成,得到預燒成物。然后,在非活性氣體氣氛中對該預燒成物進行再燒成,得到包含氧化鈰-氧化鋯系規則排列相前體、鋁酸鹽系復合氧化物前體和來自于有機物的碳化物的燒成物。這些前體和碳化物以納米水平相互分散。
固體生成物除了包含將鹽分解而成的化合物以外,還包含用于將包含鹽的水溶液凝膠化而使用的羥基羧酸。通過預燒成,將該羥基羧酸脫水,使脫水了的羥基羧酸作為有機物殘留在預燒成物中。
預燒成溫度只要是能夠將羥基羧酸脫水的溫度就不特別限定。在羥基羧酸為檸檬酸的情況下,關于預燒成溫度,優選為150~300℃。如果預燒成溫度為150℃以上,則能夠將檸檬酸脫水。更優選為175℃以上。另一方面,如果預燒成溫度為300℃以下,則脫水了的檸檬酸不會分解而形成二氧化碳等。更優選為250℃以下。進一步優選為200℃以下。在酒石酸和乳酸的情況下也可以同樣。
預燒成氣氛只要是能夠將羥基羧酸脫水的氣氛就不特別限定,例如可舉出大氣。
預燒成時間可根據固體生成物的量來適當決定。預燒成時間可以為10分鐘以上、20分鐘以上、25分鐘以上,也可以為60分鐘以下、45分鐘以下或35分鐘以下。
對如此得到的預燒成物在非活性氣體氣氛下進行再燒成。通過再燒成,由該預燒成物得到氧化鈰-氧化鋯系規則排列相前體和鋁酸鹽系復合氧化物前體。另外,將殘留在預燒成物中的有機物碳化,得到來自于有機物的碳化物。即,通過再燒成,得到包含氧化鈰-氧化鋯系規則排列相前體、鋁酸鹽系復合氧化物前體和來自于有機物的碳化物的燒成物。
再燒成溫度只要是能夠形成前體并將有機物碳化的溫度就不特別限定。關于再燒成溫度,優選為550~850℃。如果再燒成溫度為550℃以上,則可將有機物碳化。更優選為575℃以上。另一方面,如果燒成溫度為850℃以下,則生成的前體不易粒生長。更優選為800℃以下。進一步優選為700℃以下。
再燒成在非活性氣體氣氛中進行。由此,防止來自于有機物的碳化物燃燒而形成二氧化碳等。再燒成中的非活性氣體除了氬氣體等18族元素的氣體以外,還包括氮氣體等。
再燒成時間可根據預燒成物的量來適當決定。燒成時間可以為0.5小時以上、1小時以上或1.5小時以上,可以為8小時以下、4小時以下或2.5小時以下。
(燒成物的還原熱處理)
在此,關于燒成物的還原熱處理,雖然示出了實施方式1和實施方式2,但不受它們所限定。
<實施方式1>
在本發明的一個實施方式中,對包含氧化鈰-氧化鋯系規則排列相前體和鋁酸鹽系復合氧化物前體的燒成物進行還原熱處理,得到燒綠石相和鋁酸鹽系復合氧化物相互分散的第1復合體。
通常,對氧化鈰和氧化鋯的固溶體進行還原熱處理時,可得到燒綠石相。但是,在還原熱處理中,燒綠石相進行粒生長。在用鑭置換鈰的一部分、用釔置換鋯的一部分的情況下也同樣。
另一方面,在本發明的制造方法中,對包含氧化鈰-氧化鋯系規則排列相前體和鋁酸鹽系復合氧化物前體的燒成物進行還原熱處理。這樣,在一邊將氧化鈰-氧化鋯系規則排列相前體還原一邊生成燒綠石相的同時,使鋁酸鹽系復合氧化物生成。此時,鋁酸鹽系復合氧化物成為擴散阻擋材料,抑制燒綠石相進行粒生長。另外,由于氧化鈰-氧化鋯系規則排列相前體和鋁酸鹽系復合氧化物前體以納米水平相互分散,因此燒綠石相和鋁酸鹽系復合氧化物也以納米水平相互分散,從而形成第1復合體。
在對除了包含氧化鈰-氧化鋯系規則排列相前體和鋁酸鹽系復合氧化物前體以外、還包含來自于有機物的碳化物的燒成物進行還原熱處理的情況下,鋁酸鹽系復合氧化物和來自于有機物的碳化物成為擴散阻擋材料,抑制燒綠石相進行粒生長。另外,由于氧化鈰-氧化鋯系規則排列相前體、鋁酸鹽系復合氧化物前體和來自于有機物的碳化物以納米水平相互分散,因此燒綠石相、鋁酸鹽系復合氧化物和來自于有機物的碳化物也以納米水平相互分散,從而形成第1復合體。進一步地,由于來自于有機物的碳化物也抑制燒綠石相和鋁酸鹽系復合氧化物無間隙地凝集,因此第1復合體的比表面積也提高。
如上所述,氧化鈰-氧化鋯系規則排列相前體具有鈰、鋯和氧不規則排列的結構。另外,鋁酸鹽系復合氧化物前體具有不完全的鋁酸鹽系復合氧化物的結構。關于還原熱處理溫度,只要是適合于還原氣氛穩定地作用于這些前體中的氧、從而使不規則的排列或不完全的結構成為規則的排列或完全的結構的溫度就不特別限定。
作為目標,關于還原熱處理溫度,優選為900~1350℃。如果還原熱處理溫度為900℃以上,則還原性氣氛也容易作用于前體內部的氧。更優選為950℃以上,進一步優選為1000℃以上。另一方面,如果還原熱處理溫度為1350℃以下,則還原性氣氛不會過度作用于前體中的氧,由此難以產生阻礙鋁酸鹽系復合氧化物和/或來自于有機物的碳化物作為擴散阻擋材料發揮作用的事情。更優選為1325℃以下,進一步優選為1200℃以下。
還原熱處理時間可根據前體的量來適當決定。還原熱處理時間可以為0.5小時以上、1.5小時以上或2.5小時以上,可以為8小時以下、6小時以下或3.5小時以下。
還原熱處理的氣氛只要是還原氣氛就不特別限定,但例如可舉出CO、HC、H2和其它烴氣體等。也可以是這些氣體與氬氣體等的混合氣體。
<實施方式2>
在實施方式2中,還原熱處理也可以包括如下。將羥基羧酸和水添加至包含氧化鈰-氧化鋯系規則排列相前體和鋁酸鹽系復合氧化物前體的燒成物,得到混合溶液。接著,使混合溶液干燥,得到干燥體。然后,在使該干燥體中的有機物殘留的溫度下對干燥體進行準燒成,得到準燒成物。進而,在非活性氣體氣氛中對準燒成物進行再燒成,得到包含氧化鈰-氧化鋯系規則排列相前體、鋁酸鹽系復合氧化物前體和來自于有機物的碳化物的再燒成物,將該再燒成物供給上述的還原熱處理。
添加至燒成物的羥基羧酸可以與用于生成凝膠而使用的羥基羧酸同樣。
添加至燒成物的羥基羧酸在準燒成物中作為有機物殘留,在再燒成后成為來自于有機物的碳化物。羥基羧酸的量可根據使來自于有機物的碳化物生成的量來適當決定。
關于羥基羧酸的質量,在將燒成物的質量設為1時,優選為4~12(質量比)。如果羥基羧酸的質量比為4以上,則來自于有機物的碳化物的作用顯著呈現。更優選為6以上。另一方面,如果羥基羧酸的質量比為12以下,則在氧化熱處理后,來自于有機物的碳化物大量殘留,由此不會對氧吸留材料的性能產生不良影響。更優選為10以下。
關于混合溶液的干燥,只要是直至不對干燥體的準燒成產生不良影響的程度來使干燥體中的水分蒸發即可。
關于干燥體的準燒成,可以與固體生成物的預燒成同樣。但是,與固體生成物的情形相比,在干燥體的情況下,羥基羧酸沒有滲透至干燥體的內部。因此,也可以在準燒成的前半程,在低溫下使羥基羧酸滲透到干燥體中,在準燒成的后半程,在高溫下使滲透到了干燥體中的羥基羧酸全部脫水。例如,在準燒成的前半程,在150~200℃下持續加熱干燥體15~45分鐘,在準燒成的后半程,在250~350℃下持續加熱干燥體5~15分鐘。
準燒成物的再燒成可以與預燒成物的再燒成同樣。
通過再燒成得到的再燒成物包含氧化鈰-氧化鋯系規則排列相前體、鋁酸鹽系復合氧化物前體和來自于有機物的碳化物。如果將如此得到的再燒成物供給上述的還原熱處理,則能夠使鋁酸鹽系復合氧化物和來自于有機物的碳化物成為擴散阻擋材料。而且,氧化鈰-氧化鋯系規則排列相和鋁酸鹽系復合氧化物以納米水平相互分散。
(第1復合體的氧化熱處理)
對第1復合體進行氧化熱處理,得到使燒綠石相的至少一部分成為κ相的第2分散相。
關于氧化鈰-氧化鋯系規則排列相,存在還原狀態的燒綠石相和氧化狀態的κ相。
由于第1復合體是對氧化鈰-氧化鋯系規則排列相前體進行還原熱處理而得到的,因此第1復合體中的氧化鈰-氧化鋯系復合氧化物具有還原狀態的燒綠石相。因此,通過對第1復合體進行氧化熱處理,使燒綠石相變成κ相,形成第2復合體。使第1復合體中的全部燒綠石相成為κ相是理想的,但使燒綠石相的至少一部分成為κ相即可。
另外,通過對第1復合體進行氧化熱處理,使第1復合體中殘留的來自于有機物的碳化物成為二氧化碳等,從而能夠從第1復合體中除去碳化物。
氧化熱處理的溫度只要能夠使燒綠石相成為κ相、使第1復合體中殘留的來自于有機物的碳化物燃燒以形成二氧化碳等就不特別限定。氧化熱處理溫度優選為400~700℃。如果氧化熱處理溫度為400℃以上,則能夠使大部分燒綠石相成為κ相,并且能夠使第1復合體中的大部分碳化物燃燒。更優選為450℃以上。進一步優選為500℃以上。另一方面,如果氧化熱處理溫度為700℃以下,則第2復合體中的燒綠石相、κ相和鋁酸鹽系復合氧化物不會進行粒生長。更優選為650℃以下,進一步優選為600℃以下。
氧化熱處理時間可根據第1復合體的量來適當決定。氧化熱處理時間可以為0.5小時以上、1小時以上或1.5小時以上,可以為8小時以下、4小時以下或2.5小時以下。
氧化熱處理的氣氛只要能夠將第1復合體中的燒綠石相的至少一部分氧化以形成κ相就不特別限定,例如可舉出大氣。
在對第1復合體進行氧化熱處理后的第2復合體中,氧化鈰-氧化鋯系規則排列相和鋁酸鹽系復合氧化物以納米水平相互分散。
圖1是示出通過本發明的制造方法得到的氧吸留材料的組織的示意圖。氧化鈰-氧化鋯系規則排列相在氧化狀態下為κ相,在還原狀態下為燒綠石相。如圖1所示,在大氣氣氛中,氧化鈰-氧化鋯系規則排列相1具有κ相1a和燒綠石相1b。氧化鈰-氧化鋯系規則排列相1和鋁酸鹽系復合氧化物2以納米水平相互分散。
氧化鈰-氧化鋯系規則排列相1和鋁酸鹽系復合氧化物2各自可以為粒子。另外,κ相1a和/或燒綠石相1b可以為粒子。即,氧化鈰-氧化鋯系規則排列相1可以是氧化鈰-氧化鋯系規則排列相粒子,κ相1a可以是κ相粒子,燒綠石相1b可以是燒綠石相粒子,而且,鋁酸鹽系復合氧化物2可以是鋁酸鹽系復合氧化物粒子。
也可以殘留沒有從前體變為氧化鈰-氧化鋯系規則排列相1的非規則相(隨機相)(未圖示)。非規則相可以為非規則相粒子。
(再氧化熱處理)
在氧化熱處理后,根據需要也可以進一步進行再氧化熱處理。通過再氧化熱處理,氧吸留材料的H2消耗量改善。
再氧化熱處理溫度只要是能夠使氧吸留材料的H2消耗量改善的溫度就不特別限定,但典型地為高于氧化熱處理溫度的溫度。
關于再氧化熱處理溫度,優選為850~1150℃。如果再氧化熱處理溫度為850℃以上,則能夠使氧吸留材料的H2消耗量改善。更優選為900℃以上。進一步優選為950℃以上。另一方面,如果再氧化熱處理溫度為1150℃以下,則不會導致粒子彼此燒結。更優選為1100℃以下。進一步優選為1050℃以下。在此,粒子是指氧化鈰-氧化鋯系規則排列相1和鋁酸鹽系復合氧化物2的粒子。
再氧化熱處理時間可根據氧化熱處理后的第2復合體的量來適當決定。再氧化熱處理時間可以為3小時以上、4小時以上或4.5小時以上,可以為7小時以下、6小時以下或5.5小時以下。
(催化劑金屬微粒的載持)
通過目前為止說明的制造方法得到的氧吸留材料(第2復合體)可在其上載持能夠作為催化劑使用金屬微粒,從而制成廢氣凈化用催化劑。
作為金屬微粒,例如可以以約0.01~5.0質量%載持粒徑2~6nm的釕(Ru)、銠(Rh)、鈀(Pd)、鋨(Os)、銥(Ir)、鉑(Pt)、金(Au)、銅(Cu)、鐵(Fe)、鎳(Ni)等金屬、其氧化物或它們的任意組合。
作為金屬微粒的載持方法,不特別限定,例如可舉出含浸載持法和表面析出法等,可使用一般的方法。
(氧吸留材料)
接著,對通過目前為止說明的制造方法制造的氧吸留材料進行說明。
(相互分散)
在通過本發明的制造方法制造的氧吸留材料中,氧化鈰-氧化鋯系規則排列相和鋁酸鹽系復合氧化物以納米水平相互分散。
在氧化鈰-氧化鋯系規則排列相中存在還原狀態的燒綠石相(Ce2Zr2O7)和氧化狀態的κ相(Ce2Zr2O8)。在氧化鈰-氧化鋯系規則排列相的全部為還原狀態的情況下,由Ce2Zr2O7表示,在氧化鈰-氧化鋯系規則排列相的全部為氧化狀態的情況下,由Ce2Zr2O8表示。在還原狀態和氧化狀態混合存在的情況下,Ce2Zr2Ox的x為超過7且小于8。另外,Ce的一部分也可以被La置換。
在通過本發明的制造方法制造的氧吸留材料中,氧化鈰-氧化鋯系規則排列相的至少一部分為κ相,該氧化鈰-氧化鋯系規則排列相整體由Ce2Zr2Ox(其中x超過7且8以下)表示。
鋁酸鹽系復合氧化物只要成為氧化鈰-氧化鋯系規則排列相的粒生長的擴散阻擋材料就不特別限定。例如可舉出MgAl2O4、LaAlO3和CaAl4O7。
(氧化鈰-氧化鋯系規則排列相的粒徑)
通過氧化鈰-氧化鋯系規則排列相和鋁酸鹽系復合氧化物的納米水平的相互分散,能夠抑制氧化鈰-氧化鋯系規則排列相的粒生長,因此氧化鈰-氧化鋯系規則排列相的粒徑以(222)平均粒徑計為18~33nm。
如下那樣定義(222)平均粒徑。對氧吸留材料取得X射線衍射譜,進而對氧化鈰-氧化鋯系規則排列相的(111)衍射線和(222)衍射線進行精密分析,從(222)衍射線的半峰值求出與氧化鈰-氧化鋯系規則排列相的(222)垂直的方向的微晶粒徑,將其作為(222)平均粒徑。
如果(222)平均粒徑為33nm以下,則可顯著地確認氧化鈰-氧化鋯系規則排列相和鋁酸鹽系復合氧化物在納米水平的相互分散效果。如果(222)平均粒徑為25nm以下,則可特別顯著地確認該效果。
另一方面,(222)平均粒徑可以為18nm以上。(222)平均粒徑與由(鋁酸鹽系復合氧化物的摩爾數)/(氧化鈰-氧化鋯系規則排列相的摩爾數)表示的摩爾比存在關聯。如果(222)平均粒徑為18nm以上,則在享有明顯的粒生長抑制作用的同時,在氧吸留材料中氧化鈰-氧化鋯系復合氧化物的相對量不過度減少。
實施例
以下,通過實施例和比較例進一步具體說明本發明。予以說明,本發明不受它們所限定。
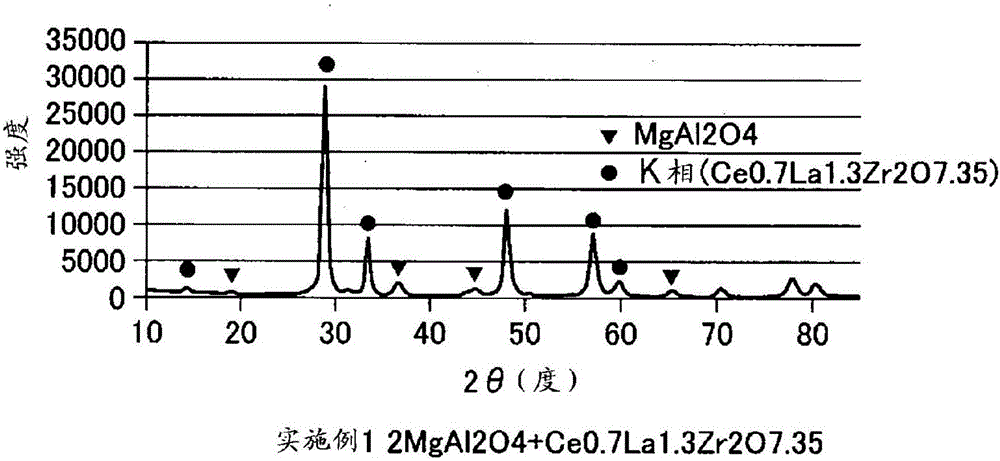
(實施例1)
實施例1為包括上述的<實施方式A>和<實施方式1>的例子。以下示出詳細內容。
將Mg(NO3)2·6H2O:10mmol、Al(NO3)3·9H2O:20mmol、Ce(NO3)3·6H2O:3.5mmol、La(NO3)3·6H2O:6.5mmol和ZrO(NO3)2·H2O:10mmol加入50mL的純水中并攪拌,溶解,制成金屬離子溶液。將檸檬酸一水合物:50mmol加入10mL的純水中并攪拌,溶解,制成檸檬酸溶液。將金屬離子溶液加入檸檬酸溶液中并攪拌,制成混合液。一邊攪拌混合液一邊加熱至80℃,使水分蒸發。在大量水分蒸發、粘性因檸檬酸的氫鍵而變高的階段,將其裝入真空干燥器,在60℃下干燥約10小時,得到凝膠。
用加熱器將凝膠在180℃下持續加熱10~30分鐘,將硝酸鹽分解。然后,將分解后的固體生成物裝入坩堝,使用電爐在200℃下持續加熱30分鐘,使固體生成物干燥。
將干燥的固體生成物在大氣中于750℃下持續燒成3小時,得到包含氧化鈰-氧化鋯系規則排列相前體和鋁酸鹽系復合氧化物前體的燒成物。
將該燒成物在5%H2-Ar氣體流通下于1100℃下持續還原熱處理3小時,得到第1復合體。
進而,將第1復合體在大氣中于550℃下持續氧化熱處理2小時,得到第2復合體。通過該氧化熱處理,第1復合體中的燒綠石相的至少一部分成為κ相。這樣,得到實施例1的氧吸留材料。予以說明,在實施例1的氧吸留材料的制作時,以由(鋁酸鹽系復合氧化物的摩爾數)/(氧化鈰-氧化鋯系規則排列相的摩爾數)表示的摩爾比成為2的方式設定Mg(NO3)2·6H2O、Al(NO3)3·9H2O、Ce(NO3)3·6H2O、La(NO3)3·6H2O和ZrO(NO3)2·H2O的進料量。
(實施例2~5)
以上述摩爾比成為0.5、0.75、1.0和3.0的方式改變Mg(NO3)2·6H2O、Al(NO3)3·9H2O、Ce(NO3)3·6H2O、La(NO3)3·6H2O和ZrO(NO3)2·H2O的進料量,除此以外,與實施例1同樣地操作,分別制作實施例2、實施例3、實施例4和實施例5的氧吸留材料。
(實施例6)
將Al(NO3)3·9H2O:10mmol、Ce(NO3)3·6H2O:6mmol、La(NO3)3·6H2O:14mmol和ZrO(NO3)2·H2O:10mmol加入50mL的純水中并攪拌,溶解,制成金屬離子溶液,將檸檬酸一水合物:40mmol加入10mL的純水中并攪拌,溶解,制成檸檬酸溶液,除此以外,與實施例1同樣地操作,得到實施例6的氧吸留材料。
(實施例7)
將Ca(NO3)2·4H2O:5mmol、Al(NO3)3·9H2O:20mmol、Ce(NO3)3·6H2O:5mmol和ZrO(NO3)2·H2O:5mmol加入50mL的純水中并攪拌,溶解,制成金屬離子溶液,將檸檬酸一水合物:35mmol加入10mL的純水中并攪拌,溶解,制成檸檬酸溶液,除此以外,與實施例1同樣地操作,得到實施例7的氧吸留材料。
(實施例8)
將Mg(NO3)2·4H2O:5mmol、Al(NO3)3·9H2O:10mmol、Ce(NO3)3·6H2O:5mmol和ZrO(NO3)2·H2O:5mmol加入50mL的純水中并攪拌,溶解,制成金屬離子溶液,將檸檬酸一水合物:35mmol加入10mL的純水中并攪拌,溶解,制成檸檬酸溶液,除此以外,與實施例1同樣地操作,得到實施例8的氧吸留材料。
(實施例9)
實施例9為包括上述的<實施方式B>和<實施方式1>的例子。以下示出詳細內容。
將Mg(NO3)2·6H2O:10mmol、Al(NO3)3·9H2O:20mmol、Ce(NO3)3·6H2O:3.5mmol、La(NO3)3·6H2O:6.5mmol和ZrO(NO3)2·H2O:10mmol加入50mL的純水中并攪拌,溶解,制成金屬離子溶液。將檸檬酸一水合物:50mmol加入10mL的純水中并攪拌,溶解,制成檸檬酸溶液。將金屬離子溶液加入檸檬酸溶液中并攪拌,制成混合液。一邊攪拌混合液一邊加熱至80℃,使水分蒸發。在大量水分蒸發、粘性因檸檬酸的氫鍵而變高的階段,將其裝入真空干燥器,在60℃下干燥約10小時,得到凝膠。
用加熱器將凝膠在180℃下持續加熱10~30分鐘,將硝酸鹽分解。然后,將分解后的固體生成物裝入坩堝,使用電爐在200℃下持續加熱30分鐘,進行預燒成,得到殘留有脫水的檸檬酸的預燒成物。
將預燒成物在Ar氣體氣氛中于600℃下持續再燒成3小時,得到包含氧化鈰-氧化鋯系規則排列相前體、鋁酸鹽系復合氧化物前體和來自于脫水的檸檬酸的碳化物的燒成物。
將該燒成物在Ar氣體流通下于1100℃下持續還原熱處理3小時,得到第1復合體。
進而,將第1復合體在大氣中于600℃下持續氧化熱處理2小時,得到第2復合體,作為實施例9的氧吸留材料。予以說明,通過該氧化熱處理,第1復合體中的來自于脫水的檸檬酸的碳化物燃燒,在第2復合體中沒有碳化物。另外,通過該氧化熱處理,第1復合體中的燒綠石相的至少一部分成為κ相。
(實施例10)
將實施例1的氧吸留材料進一步在大氣中于1000℃下持續再氧化熱處理5小時,得到實施例10的氧吸留材料。
(實施例11)
實施例11為包括上述的<實施方式A>和<實施方式2>的例子。以下示出詳細內容。
將Mg(NO3)2·6H2O:10mmol、Al(NO3)3·9H2O:20mmol、Ce(NO3)3·6H2O:3.5mmol、La(NO3)3·6H2O:6.5mmol和ZrO(NO3)2·H2O:10mmol加入50mL的純水中并攪拌,溶解,制成金屬離子溶液。將檸檬酸一水合物:50mmol加入10mL的純水中并攪拌,溶解,制成檸檬酸溶液。將金屬離子溶液加入檸檬酸溶液中并攪拌,制成混合液。一邊攪拌混合液一邊加熱至80℃,使水分蒸發。在大量水分蒸發、粘性因檸檬酸的氫鍵而變高的階段,將其裝入真空干燥器,在60℃下干燥約10小時,得到凝膠。
用加熱器將凝膠在200℃下持續加熱30分鐘,將硝酸鹽分解,同時使硝酸鹽分解后的固體生成物干燥。
將干燥的固體生成物在大氣中于400℃下持續燒成1小時,得到包含氧化鈰-氧化鋯系規則排列相前體和鋁酸鹽系復合氧化物前體的燒成物。
向燒成物添加檸檬酸水溶液,得到混合溶液。燒成物的質量與檸檬酸的質量設為1:6。然后,使混合溶液中的水分蒸發,得到干燥體。
對干燥體進行準燒成。準燒成分為在200℃下30分鐘和300℃下10分鐘兩次來進行。通過該準燒成,將干燥體中的檸檬酸脫水,使脫水的檸檬酸作為有機物殘留在準燒成物中。
將準燒成物在Ar氣體氣氛中于600℃下持續再燒成3小時,得到包含氧化鈰-氧化鋯系規則排列相前體、鋁酸鹽系復合氧化物前體和來自于脫水的檸檬酸的碳化物的再燒成物。
將該再燒成物在Ar氣體流通下于1100℃下持續還原熱處理3小時,得到第1復合體。
進而,將第1復合體在大氣中于550℃下持續氧化熱處理2小時,得到第2復合體,作為實施例11的氧吸留材料。通過該氧化熱處理,第1復合體中的燒綠石相的至少一部分成為κ相。
(實施例12)
將金屬離子溶液以進料組成計設為2MgAlO4+CeLaY0.2Zr2,除此以外,與實施例11同樣地操作,得到實施例12的氧吸留材料。予以說明,在如實施例11那樣配合硝酸鹽時,金屬離子溶液的進料組成為2MgAlO4+La1.3Y0.2Zr2。
(實施例13)
將干燥的固體生成物在大氣中于600℃下持續燒成3小時,得到包含氧化鈰-氧化鋯系規則排列相前體和鋁酸鹽系復合氧化物前體的燒成物,除此以外,與實施例11同樣地操作,得到實施例13的氧吸留材料。
(實施例14)
將燒成物的質量與檸檬酸的質量設為1:10,且將干燥的固體生成物在大氣中于600℃下持續燒成3小時,得到包含氧化鈰-氧化鋯系規則排列相前體和鋁酸鹽系復合氧化物前體的燒成物,除此以外,與實施例12同樣地操作,得到實施例14的氧吸留材料。
(實施例15)
將實施例1的還原熱處理溫度設為1300℃,除此以外,與實施例1同樣地操作,得到實施例15的氧吸留材料。
(實施例16)
將1mg的鉑(Pt)載持于8.63g(10mmol)的實施例9的氧吸留材料。然后,將載持有該鉑的氧吸留材料與實施例10的氧吸留材料8.63g(10mmol)物理混合,制成廢氣凈化用催化劑。
在實施例9的氧吸留材料上載持鉑的方法如下所述。使規定量的氧吸留材料的粉末分散在100mL的純水中,進行攪拌。在該溶液中加入相當于鉑1mg的份量的二硝基二氨合鉑溶液,在熱攪拌器上進行攪拌加熱。其后,將攪拌加熱了的溶液蒸發干固。使其在120℃下進一步持續干燥2小時,接著,在500℃持續燒成2小時,制成鉑催化劑。
關于物理混合方法,使用粉體混合器,將鉑催化劑和實施例10的氧吸留材料的粉末混合之后,進一步用研缽將其混合,制成廢氣凈化用催化劑。
(比較例1)
將Ce(NO3)3·6H2O:7.9mmol、La(NO3)3·6H2O:14.6mmol和ZrO(NO3)2·H2O:22.5mmol加入30mL的純水中并攪拌,溶解,制成金屬離子溶液。將檸檬酸一水合物:45mmol加入10mL的純水中并攪拌,溶解,制成檸檬酸溶液。將金屬離子溶液加入檸檬酸溶液中并攪拌,制成混合液。一邊攪拌混合液一邊加熱至80℃,使水分蒸發。在大量水分蒸發、粘性因檸檬酸的氫鍵而變高的階段,將其裝入真空干燥器,在60℃下干燥約10小時,得到凝膠。
用加熱器將凝膠在180℃下持續加熱10~30分鐘,將硝酸鹽分解。然后,將分解后的固體生成物裝入坩堝,使用電爐在200℃下持續加熱30分鐘,使固體生成物干燥。
將干燥的固體生成物在大氣中于750℃下持續燒成3小時,得到包含氧化鈰-氧化鋯系規則排列相前體的燒成物。
將該燒成物在5%H2-Ar氣體流通下于1100℃下持續還原熱處理3小時,得到燒綠石相。
進而,將該燒綠石相在大氣中于550℃下持續氧化熱處理2小時,得到燒綠石相的至少一部分成為κ相的氧吸留材料(Ce0.7La1.3Zr2Ox)。
(比較例2)
將Ce(NO3)3·6H2O:2.5mmol、La(NO3)3·6H2O:7.5mmol和ZrO(NO3)2·H2O:10mmol加入30mL的純水中并攪拌,溶解,制成金屬離子溶液,將檸檬酸一水合物:20mmol加入10mL的純水中并攪拌,溶解,制成檸檬酸溶液,除此以外,與比較例1同樣地操作,得到氧吸留材料(Ce0.5La1.5Zr2O7.25)。
(比較例3)
將Ce(NO3)3·6H2O:10mmol和ZrO(NO3)2·H2O:10mmol加入20mL的純水中并攪拌,溶解,制成金屬離子溶液,將檸檬酸一水合物:20mmol加入10mL的純水中并攪拌,溶解,制成檸檬酸溶液,除此以外,與比較例1同樣地操作,得到氧吸留材料(Ce2Zr2O8)。
(比較例4)
將比較例1中得到的氧吸留材料進一步在大氣中于1000℃下持續再氧化熱處理5小時,得到比較例4的氧吸留材料。
(比較例5)
以碳作為還原劑,將具有Ce1.84Zr2.16Ox的組成的氧化鈰與氧化鋯的固溶體在1700℃下進行還原熱處理,得到具有微晶為微米水平的氧化鈰-氧化鋯系規則排列相的氧吸留材料。
(比較例6)
將具有Ce0.7La1.3Zr2Ox的組成的氧化鈰與氧化鋯的固溶體在5%H2-Ar氣體流通下于1300℃下持續還原熱處理3小時,得到具有氧化鈰-氧化鋯系規則排列相的氧吸留材料。
(比較例7)
將1mg的鉑載持于具有約100m2/g的表面積的氧化鋁(Al2O3)4.08g(40mmol)。然后,將載持有該鉑的氧吸留材料與比較例1的氧吸留材料57g(20mmol)物理混合,制成廢氣凈化用催化劑。鉑載持方法和物理混合方法與實施例16同樣。
(評價)
按如下評價如此得到的實施例1~實施例16和比較例1~7的氧吸留材料。
(X射線衍射分析)
對實施例1和實施例6~9以及比較例2~3的氧吸留材料進行X射線衍射分析。在圖2A~圖2E中示出實施例1和實施例6~9的衍射譜。另外,根據圖2的各衍射譜與JCPD卡片的對比,確定各氧吸留材料的組成。將結果示于表1。
表1
接著,對圖2A~圖2D的氧化鈰-氧化鋯系規則排列相的(111)衍射線和(222)衍射線進行精密測定,對于各氧吸留材料,從(222)衍射線的半峰值求出(222)平均粒徑。將結果示于圖3。
由表1和圖3可知,與比較例2和比較例3的(222)平均粒徑相比,實施例1和實施例6~8的(222)平均粒徑小。由于在比較例2和比較例3的氧吸留材料中,氧化鈰-氧化鋯系規則排列相單獨存在,因此在實施例1和實施例6~8的氧吸留材料中,可確認鋁酸鹽系復合氧化物成為氧化鈰-氧化鋯系規則排列相的粒生長的擴散阻擋材料。
另外,在表2和圖4中示出在實施例1~實施例5中使由(鋁酸鹽系復合氧化物的摩爾數)/(氧化鈰-氧化鋯系規則排列相的摩爾數)表示的摩爾比(以下有時簡單稱作“摩爾比”)以0.5~3變化的氧吸留材料的(222)平均粒徑。在表2和圖4中,還一并示出了比較例1(摩爾比相當于0)的氧吸留材料的(222)平均粒徑。
表2
根據表2和圖4,可確認摩爾比為0.5以上時,鋁酸鹽系復合氧化物作為擴散阻擋材料顯著地發揮作用。進而,可確認在摩爾比為1以上時,鋁酸鹽系復合氧化物作為擴散阻擋材料特別顯著地發揮作用。
此外,測定了實施例1和實施例10以及比較例1和比較例4的(222)平均粒徑。將結果示于表3。
表3
實施例10是將實施例1的氧吸留材料進行再氧化熱處理而得到的。由表3可知,通過再氧化熱處理,(222)平均粒徑沒有大的變化。但是,關于后述的H2消耗量,實施例10超過實施例1。
另外,關于各氧吸留材料,求出作為表示氧化鈰-氧化鋯系規則排列相的生成狀態的一項指標的I值。在X射線衍射譜中,2θ=14.5°的衍射線為歸屬于κ相的(111)面的衍射線,2θ=29°的衍射線為歸屬于κ相的(222)面的衍射線,與歸屬于氧化鈰-氧化鋯固溶體(CZ固溶體)的(111)面的衍射線重合。因此,可將作為兩者的衍射線的強度比的I值設為表示κ相的維持率(存在率)的指標。即,通過I值={(111)峰的面積}/{(222)峰的面積}求出I值,作為表示κ相的維持率(存在率)的指標。將I值的計算結果示于圖5。
由圖5可知,沒有用La置換Ce的實施例7和實施例8以及比較例3的I值均為同程度的值。由此可認為,在實施例7和實施例8以及比較例3的任一者中,雖然平均粒徑不同,但都生成了氧化鈰-氧化鋯系規則排列相。因此可認為,后述的氧吸留能力等特性的不同歸因于氧化鈰-氧化鋯系規則排列相的粒徑的不同。
予以說明,用La置換了Ce的實施例1和實施例6以及比較例2的I值低于實施例7和實施例8以及比較例3的I值。認為這是由于在實施例1和實施例6以及比較例2中,Ce4+被La3+置換,因此氧化鈰-氧化鋯系規則排列相即使為κ相,該氧化鈰-氧化鋯系規則排列相也呈現燒綠石相的形態(Haruo Kishimoto等,Journal of Alloys and Compounds,312(2000),pp.94-103)。但是,置換了La的實施例1和實施例6以及比較例2顯示同程度的I值。由此,關于它們,也可確認生成了氧化鈰-氧化鋯系規則排列相。因此可認為,后述的氧吸留能力等特性的不同歸因于粒徑的不同。
另外,同樣地,關于實施例11~實施例14的氧吸留材料,也求出(222)平均粒徑。將結果示于表4。在表4中,一并示出金屬離子溶液的進料組成和加入燒成物的檸檬酸的量(質量比)。
表4
由表4可知,確認了實施例11~實施例14中的任一者的氧吸留材料具有與其它實施例同等的(222)平均粒徑。由此可確認,即使將檸檬酸加至包含氧化鈰-氧化鋯系規則排列相前體和鋁酸鹽系復合氧化物前體的燒成物,也會得到在還原熱處理時成為阻擋材料的來自于脫水的檸檬酸的碳化物。
(透射電子顯微鏡觀察和分析)
用透射電子顯微鏡(TEM:Transmission Electron Microscopy)觀察實施例8的氧吸留材料,基于其觀察結果,分析氧吸留材料的晶體結構。
圖6是示出在對實施例8的氧吸留材料進行透射電子顯微鏡觀察的結果(晶格像)中其第1視場的圖。圖7A是示出由圖6的(a)所示區域的FFT(快速傅立葉變換)譜和基于其的衍射模擬結果的圖。圖7B是示出由圖6的(b)所示區域的FFT(快速傅立葉變換)譜和基于其的衍射模擬結果的圖。圖7C是示出由圖6的(c)所示區域的FFT(快速傅立葉變換)譜和基于其的衍射模擬結果的圖。
圖8是示出在對實施例8的氧吸留材料進行電子顯微鏡觀察的結果(晶格像)中第2視場的圖。圖9是示出由圖8的(a)所示區域的FFT(快速傅立葉變換)譜和基于其的衍射模擬結果的圖。
予以說明,基于FFT譜的衍射模擬是指用軟件(ReciPro)計算出假設的晶體的電子束衍射譜,將該計算結果與實測的電子束衍射譜比較來確定晶體結構。
圖7A示出電子束的入射方位為[1-10]時的晶體結構,由該晶體結構可知,圖6的區域(a)為Mg2Al2O4。圖7B示出電子束的入射方位為[0-11]時的晶體結構,由該晶體結構可知,圖6的區域(b)為Ce2Zr2O7。圖7C示出電子束的入射方位為[001]時的晶體結構,由該晶體結構可知,圖6的區域(c)為MgAl2O4。
圖9示出電子束的入射方位為[1-10]時的晶體結構,由該晶體結構可知,圖8的區域(a)為Mg2Al2O4。
另外,通過同樣的分析,確認了圖8的區域(b)的白線表示MgAl2O4的(111)面,其間隔為0.47nm。圖8的區域(c)的白線表示MgAl2O4的(111)面,其間隔為0.47nm。而且,圖8的區域(d)的白線表示Ce2Zr2O7的(111)面,其間隔為0.33nm。
予以說明,由于透射電子顯微鏡觀察是通過在真空中對試樣照射電子束而進行的,因此即使在成為κ相(Ce2Zr2O8)的試樣中,通過在觀察中形成真空,有時也成為燒綠石相(Ce2Zr2O7)。
如從圖6~圖9可知的那樣,確認了關于實施例8的氧吸留材料,氧化鈰-氧化鋯系規則排列相(Ce2Zr2O8和/或Ce2Zr2O7)和鋁酸鹽系復合氧化物(MgAl2O4)為8~12nm,它們以納米水平相互分散。
另外,關于實施例9的氧吸留材料,進行了基于透射電子顯微鏡觀察和電子能量損失能譜(EELS:Electron Energy Loss Spectroscopy)的映射。將結果示于圖10A~圖10C。圖10A是示出對實施例9的氧吸留材料使用透射電子顯微鏡進行觀察的透射圖像的圖。圖10B是示出對圖10A的觀察區域進行Ce-N EELS映射的結果的圖。圖10C是示出對圖10A的觀察區域進行Al-K EELS映射的結果的圖。
在圖10A中,觀察到了10nm左右的晶粒。另外,在圖10A和圖10B中,確認了氧化鈰-氧化鋯系規則排列相(Ce0.7La1.3Zr2O7.35)和鋁酸鹽系復合氧化物(MgAl2O4)以納米水平相互分散。
(比表面積)
通過BET法測定了實施例1和實施例9以及比較例1的比表面積。將結果示于表5。在表5中,還一并示出(222)平均粒徑和鋁酸鹽系復合氧化物(MgAl2O4)的平均粒徑。還從各氧吸留材料的X射線衍射譜求出了鋁酸鹽系復合氧化物的平均粒徑。
表5
如從表5可知的那樣,與比較例1相比,在實施例9中,比表面積成為約25倍。可認為這是由于在實施例9中,在為了得到第1復合體的還原熱處理時,在氧化鈰-氧化鋯系規則排列相前體(Ce0.7La1.3Zr2Ox)和鋁酸鹽系復合氧化物(MgAl2O4)前體之間存在來自于脫水的檸檬酸的碳化物,氧吸留材料的比表面積因該碳化物而增加。而且,如在<實施方式B>中說明的那樣,該碳化物的存在是基于在非活性氣體氣氛中進行再燒成,由此使預生成物中的有機物碳化而得到的。
(H2消耗量)
用H2-TPR(Temperature Programed Reduction,升溫還原)法進行實施例1和實施例10以及比較例1和比較例4的氧吸留材料的H2消耗量的測定。
利用H2-TPR法的H2消耗量的測定的條件如下所述。
測定裝置:日本ベル株式會社制催化劑分析裝置BEL-CAT
測定方法:升溫式脫離過程測定TPD(Temperature Programed Desorption)
測定單元(cell):化學吸附量測定單元
氣體流量:60mL/分鐘
檢測方法:a)熱導率檢測器(TPD)
b)四級質量分析儀(MS:Mass Spectrometry)
檢測方式:法拉第杯/二次電子倍增管
檢測極限:1ppm
氣體導入量:約1.0mL/分鐘(1大氣壓)
測定質量數:m/z;2(H2)、m/z;40(Ar)、18(H2O)
測定條件:1)室溫~500℃ 10%O2/Ar氣體流通中(升溫10℃/分鐘)
2)500℃ 10%O2/Ar氣體流通中(保持10分鐘)
3)500~40℃ 10%O2/Ar氣體流通中
4)40℃ Ar氣體流通中(保持30分鐘)
5)40~950℃ 5%H2/Ar氣體流通中(升溫速度10℃/分鐘)
6)950℃ 5%H2/Ar氣體流通中(保持20分鐘)
7)950~40℃ Ar氣體流通中
在實施例1和實施例10以及比較例1和比較例4中,如表6所示,由于氧吸留材料中的Ce含量不同,因此用Ce含量將H2消耗量歸一化,得到每單位Ce質量%的H2消耗量。
表6
將測定結果示于圖11A和圖11B。圖11A是示出實施例1和實施例10的氧吸留材料的每單位Ce質量%的H2消耗量分布的圖。圖11B是示出比較例1和比較例4的氧吸留材料的每單位Ce質量%的H2消耗量分布的圖。予以說明,雖然使用熱導率檢測器(TPD)和四級質譜儀(MS)兩者進行了測定,但由于基本上沒有確認出兩者測定結果的差異,因此示出使用熱導率檢測器(TPD)進行測定的測定結果。
圖12是示出實施例1和實施例10以及比較例1和比較例4的氧吸留材料的每單位Ce質量%的H2消耗量的圖。
如從圖11A、圖11B和圖12可知的那樣,實施例1僅室溫~950℃的H2消耗量增加,與此相對,實施例10的室溫~500℃和室溫~600℃的H2消耗量也增加。即,可確認實施例10的氧吸留材料高溫自不必說,低中溫的氧吸留能力也優異。
這是由于在實施例10中,通過對實施例1的氧吸留材料進行再氧化熱處理,將無間隙凝聚的納米水平的粒子各自分割。予以說明,粒子是指相互分散的氧化鈰-氧化鋯系規則排列相和鋁酸鹽系復合氧化物。
關于實施例1和實施例10以及比較例1和比較例4以外的氧吸留材料,也同樣地進行了H2消耗量測定,進行了各種考察。對它們的考察結果進行說明。予以說明,由于各氧吸留材料的Ce含量各自不同,因此除非另外指出,以用Ce含量歸一化的H2消耗量(每單位Ce質量%的H2消耗量)進行了考察。
圖13是示出實施例9和比較例1的氧吸留材料的每單位Ce質量%的H2消耗量分布的圖。圖14是示出實施例9和比較例1的氧吸留材料在各溫度下的每單位Ce質量%的H2消耗量的圖。
如從圖13和圖14可知的那樣,與比較例1相比,在實施例9中,從低溫到高溫,H2消耗量多。特別地,關于低中溫區域的H2消耗量,實施例9為比較例1的約2倍。即,可確認實施例9的氧吸留材料從低溫至高溫,氧吸留能力優異。
另外,圖15是示出實施例1和實施例9的氧吸留材料的H2消耗量的圖。
如從圖15可知的那樣,實施例9的氧吸留材料的H2消耗量多于實施例1。這是由于如從表5可知的那樣,實施例9的氧吸留材料的比表面積大于實施例1。實施例9的氧吸留材料的比表面積更大的原因如在表5中說明的那樣。
圖16是示出對各氧吸留材料的室溫~950℃的H2消耗量各進行了3次測定時的各次的測定結果的圖。
如從圖16可知的那樣,確認了實施例1和實施例10的氧吸留材料的H2消耗量的偏差小,能夠確保再現性。
圖17是示出實施例6和比較例5的氧吸留材料的每單位Ce質量%的H2消耗量分布的圖。
如從圖17中可知的那樣,與比較例5的氧吸留材料相比,實施例6的氧吸留材料在低中溫下的H2消耗量多。如目前為止說明的那樣,這是由于在實施例6的氧吸留材料中,氧化鈰-氧化鋯系規則排列相和鋁酸鹽系復合氧化物以納米水平相互分散。與此相對,這是由于在比較例5中,僅將碳用于將熱處理氣氛中的氧還原,碳對使氧化鈰-氧化鋯系規則排列相和鋁酸鹽系復合氧化物以納米水平相互分散不起作用。
圖18是示出實施例15和比較例6的氧吸留材料的每單位Ce質量%的H2消耗量分布的圖。
如從圖18可知的那樣,實施例15的氧吸留材料與比較例6的氧吸留材料相比,低中溫下的H2消耗量多。實施例15是在1300℃下進行還原熱處理而得到的氧吸留材料。由此確認了,即使還原熱處理溫度為1300℃,也得到了氧化鈰-氧化鋯系規則排列相和鋁酸鹽系復合氧化物以納米水平相互分散的氧吸留材料。
圖19是示出實施例16和比較例7的廢氣凈化催化劑在400℃下的每單位Ce質量%的H2消耗量的圖。
如從圖19可知的那樣,關于廢氣凈化催化劑在400℃下的每單位Ce質量%的H2消耗量,實施例16超過比較例7。由此確認了本發明的氧吸留材料能夠載持催化劑金屬來使用。
根據目前為止所說明的結果,可確認本發明的效果。
- 還沒有人留言評論。精彩留言會獲得點贊!