一種鈷鉬復合金屬多功能碳基催化劑及其制備方法和應用與流程
本發明屬于過渡金屬催化劑制備技術領域,具體涉及一種將合成氣轉化為液態烴及含氧化合物的鈷鉬復合金屬多功能碳基催化劑及其制備方法和應用。
背景技術:
隨著石油資源的日益枯竭,可再生生物質能源的利用受到多方面關注,其中以可再生生物質為原料,經氣化過程生成合成氣(CO+H2)的技術最為成熟。該過程產生的合成氣凈化后可經費托合成(FTS)或CO氫化(醇合成)。費托合成是將合成氣催化轉化為各種烴類及含氧有機化合物的過程,是理想的生產替代石油燃料的技術[Fuel Process.Technol.71(2001)149.Catal.Today 84(2003)83.]。由于碳氫化合物中的碳(C),氫(H)和氧(O)在費托合成過程中產生副產品CO2和H2O,降低了費托合成過程的總效率。保證C、H、O元素有效地轉化為醇、酯、醚、醛、酮、液態烴等最終產物不僅可以提高費托合成總效率,還能顯著提高合成氣利用率、降低氫耗量,使用適用于費托合成和加氫反應的多功能催化劑可達到這一目標。近十年來,合成氣生產汽油大多使用一步法,與兩步法和三步法相比成本更低,因而更具優勢;使用雙功能催化劑可使甲醇的合成以及甲醇合成為烴兩個反應同時進行。在相同反應介質中兩個反應的同時進行使得熱力學平衡向合成甲醇的方向移動,此外,由于酸性官能團的存在,可以實現控制產物的選擇性,這是費托合成中無法達到的。
由于過渡金屬碳化物有貴金屬催化劑一樣的活性、穩定性、廣泛選擇性,尤其是鎢和鉬的碳化物,因此在催化劑領域受到極大的關注。研究表明,碳化鉬在合成氣合成烴時非常活躍,Woo等人發現摻入K2CO3的碳化鉬能大大提高對高碳醇的選擇性;Xiang等人用K/β-Mo2C和鐵、鈷、鎳等費托合成元素整合作為催化劑,在合成烴時發現改性后的K/β-Mo2C催化劑對混合醇的合成具有更高的活性和選擇性。目前大多數研究都致力于探討碳化鉬的制備方法及催化性能,而不是含鉬的復合金屬碳化物。因此本發明提出用鈷鉬復合金屬碳基材料作為合成氣合成液態烴和含氧化合物的多功能催化劑。
制備金屬碳化物有以下幾種方法,傳統方法是高溫時將石墨碳加入金屬中直接碳化。Newsam等人最先提出兩步法制備鈷鉬雙金屬碳化物,以CoMoO4為前驅體合成了Co6Mo6C2,Co3Mo3C以及Co6Mo6C;Xiao等人提出用碳熱還原法制備鈷鉬碳化物,反應中氧化物作為前驅物并在C2H6/H2混合氣流中碳化;Liang等人用碳熱還原氫化法制備鈷鉬碳化物、納米β-Mo2C、W2C。Wang等人將過量含環六亞甲基四胺(HMT)混合鹽熱解用來制備Co3Mo3C和Co6Mo6C,其中HMT作為鉬酸鹽離子配位體和碳源參與反應。本發明采用直接碳化還原法合成鈷鉬復合金屬碳化物。
技術實現要素:
本發明的目的在于,提供一種鈷鉬復合金屬多功能碳基催化劑及其制備方法和應用,該催化劑的制備方法簡單,制得的催化劑活性高、穩定性好、選擇性高。
本發明的技術方案:一種鈷鉬復合金屬多功能碳基催化劑的制備方法,制備過程包括以下步驟:
a、催化劑前驅體的制備:將含鉬化合物溶于去離子水中,加熱至80℃,攪拌至完全溶解,再將溶液冷卻至40℃并加入含鈷化合物,再次攪拌至完全溶解;隨后將混合溶液逐滴加入盛有載體碳的燒杯中并攪拌混合,在80℃下蒸發去除溶劑,剩余固體在110℃下干燥12小時,得A品;
b、鈷鉬碳化物的制備:將步驟a中干燥所得固體A品,在氦氣流中加熱至300℃后保溫2小時,然后在惰性氣流,采用碳熱還原法或碳熱氫還原法升溫至700~1100℃,保溫2小時,最后樣品在室溫下通入含氧氣的氮氣進行2小時鈍化處理,得B品;
c、引入過渡金屬:通過浸漬法將等量或等體積過渡金屬負載于步驟b中所得B品上,隨后在80℃下蒸發去除溶劑,剩余固體在110℃下干燥12小時;或先制備納米級過渡金屬氧化物硝酸鹽、氯化物、氧化物、氫氧化物,再將其與B品均勻混和,最后將引入過渡金屬成分的B品干燥后置于氮氣流中焙燒,得C品;
d、引入堿金屬:通過浸漬法將等量或等體積的堿金屬成分負載于步驟c中制備的C品上,隨后在80℃下蒸發去除溶劑,剩余固體在110℃下干燥12小時;或先制備納米級堿金屬氧化物、氫氧化物、硝酸鹽、氯化物、碳酸鹽或硫酸鹽,再將其與步驟c中C品均勻混和,最后將引入堿金屬成分的樣品干燥后,置于350℃氮氣流中焙燒3小時,粉碎造粒即得鈷鉬復合金屬多功能碳基催化劑。
進一步地,步驟a中所述含鈷/鉬金屬化合物用量取決于鈷鉬金屬負載量,其負載量可取下列值:5wt%Co-25wt%Mo/C,10wt%Co-25wt%Mo/C,15wt%Co-25wt%Mo/C,20wt%Co-25wt%Mo/C,15wt%Co-15wt%Mo/C,15wt%Co-10wt%Mo/C和15wt%Co-5wt%Mo/C等。
進一步地,所述步驟a中含鈷化合物是氯化鈷、乙酸鈷、四水合乙酸鈷、乙酰丙酮鈷、碳酸鈷、六水合硝酸鈷、七水合硫酸鈷和四氧化三鈷中的任意一種;所述步驟a中含鉬化合物是五氯化鉬、六羰基鉬、七鉬酸銨、四氯氧化鉬、三氧化鉬和乙酸鉬中的任意一種,所述催化劑載體碳材料是活性炭、炭黑、碳納米管、碳納米纖維、富勒烯、石墨烯、石墨、生物炭或其他含碳材料。
進一步地,所述步驟c中引入的過渡金屬是鐵(Fe)、鎳(Ni)、鎢(W)、釩(V)、銅(Cu)、銀(Ag)、鉑(Pt)、鈀(Pd)、釕(Ru)、銥(Ir)、銠(Rh)中的至少一種,或過渡金屬硝酸鹽、氯化物、氧化物、氫氧化物。
進一步地,所述步驟d中引入的堿金屬為鋰、鈉、鉀、銣、銫中的至少一種,或堿金屬的氧化物、氫氧化物、硝酸鹽、氯化物、碳酸鹽或硫酸鹽,引入量為0.1~20wt%。
進一步地,所述步驟b中鈷鉬雙金屬碳化物是通過碳熱還原法(CR)或碳熱氫還原法(CHR)合成。
優選地,引入的過渡金屬量為0.1~20wt%。
優選地,所述堿金屬的氧化物為氧化鉀,所述氧化鉀的添加量為0.1~10wt%。
本發明還提供了一種鈷鉬復合金屬多功能碳基催化劑的制備方法所得的鈷鉬復合金屬多功能碳基催化劑,該鈷鉬復合金屬多功能碳基催化劑的重量比組成為:Co:Mo:C=1~50:1~50:100。
進一步地,該鈷鉬復合金屬多功能碳基催化劑的組成包括:鈷鉬復合金屬碳化物,至少一種過渡金屬和堿金屬助劑。
本發明的鈷鉬復合金屬多功能碳基催化劑在合成氣催化轉化中的應用,用于反應的合成氣是僅含有CO和H2的清潔合成氣,或是源自生物質、城市固體廢物、褐煤氣化產生的N2和CO2含量較高的合成氣。
進一步地,合成的產品是液態烴、高醇、酯、醚、醛和酮,其中,含氧化合物可達5~80%,汽油、航空燃料、煤油和柴油類液態烴可達20~95%。
按上述步驟所得鈷鉬復合金屬多功能碳基催化劑使用前須在氫氣還原氣氛下活化8小時,活化溫度400℃。
進一步地,合成液態烴、高醇、酯、醚、醛和酮過程中,合成氣轉化反應在固定床中進行,催化劑與合成氣充分接觸;反應條件為:溫度200~450℃,空速500~5000h-1,反應器壓力200~1200psi。
與現有技術相比,本發明具有如下有益效果:
本發明的催化劑是以Co、Mo為活性成分,碳為載體,用碳熱還原法(CR)或碳熱氫還原法(CHR)合成鈷鉬復合金屬碳化物,隨后利用浸漬法附加過渡金屬及堿金屬元素,然后蒸發去除溶劑,烘干造粒,得到適于合成液態烴及含氧化合物的鈷鉬復合金屬多功能碳基催化劑,該催化劑的制備方法簡單,制得的催化劑活性高、穩定性好、選擇性高。
附圖說明
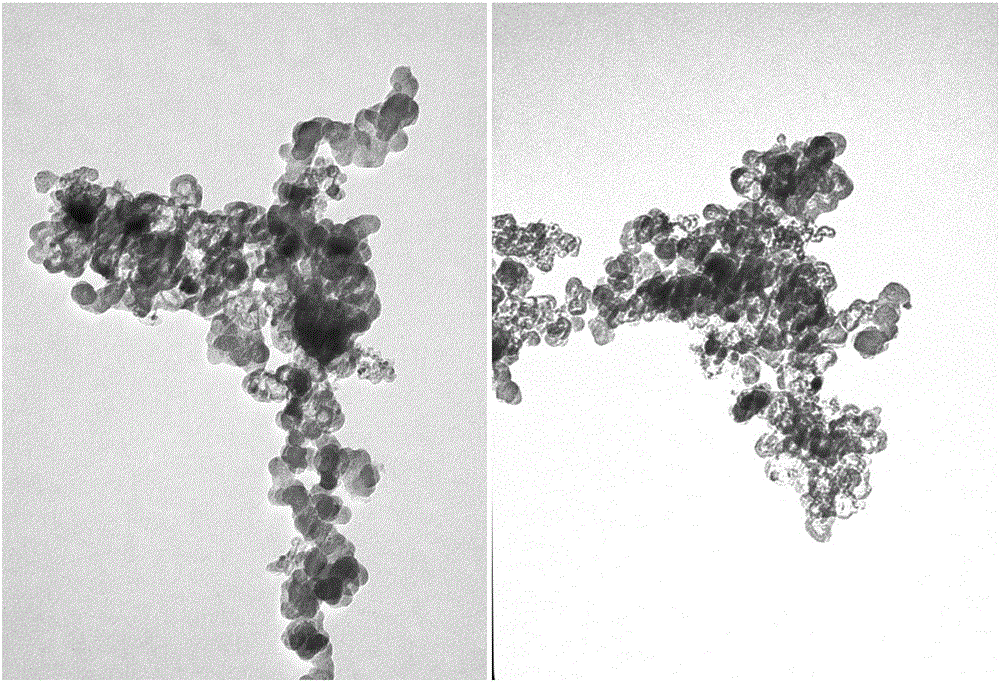
圖1是本發明實施例1中鈷鉬復合金屬多功能碳基催化劑的電鏡掃描圖(SEM);
圖2是本發明實施例1中鈷鉬復合金屬多功能碳基催化劑的透射電鏡圖(TEM)圖像;
圖3是本發明實施例6中CO轉化率、C1-C4輕質烴和液態產品選擇性隨運行時間的變化圖。
具體實施方式
下面結合附圖和實施例對本發明作進一步的說明,但并不作為對本發明限制的依據。
實施例1。一種鈷鉬復合金屬多功能碳基催化劑的制備方法,制備過程包括以下步驟:
a、用鉬酸銨((NH4)6Mo7O24·4H2O)和硝酸鈷(Co(NO3)2·6H2O)配制鈷鉬摩爾比為1:1的鹽溶液,步驟如下:7.36g鉬酸銨溶于20mL去離子水中,將該溶液加熱至80℃,攪拌至澄清液,然后再將溶液冷卻至40℃并加入12.12g硝酸鈷,再次攪拌至澄清液,隨后把制得的雙金屬鹽溶液逐滴加入盛有10g活性炭的燒杯中并不斷攪拌,并在80℃下蒸發去除溶劑,剩余固體在110℃下干燥12小時,得A品;
b、將步驟a中所得固體A品送入外徑1英寸的石英反應管,采用程序控溫加熱管式爐,樣品量約為20~200g/次,在500mL/min氦氣流中加熱至300℃,保溫2小時即得CoMoO4,然后在100~1000mL/min氦氣流或純氫氣流中以1~100℃/min的速率加熱至700~1100℃,保溫2小時,最后樣品在室溫下通入含微量氧氣的氮氣2小時進行鈍化處理,得B品;
c、取0.5g PdCl2溶于10mL去離子水,將PdCl2溶液逐滴加入步驟b所得的B品中并不斷攪拌,并在80℃下蒸發去除溶劑,得到的樣品在110℃下干燥12小時,得C品;
d、取0.5g KOH溶于10mL去離子水,將KOH溶液逐滴加入步驟c所得C品中并不斷攪拌,并在80℃下蒸發去除溶劑,得到的樣品在110℃下干燥12小時,最后將樣品置于350℃氮氣流中焙燒3小時,粉碎造粒即得鈷鉬復合金屬多功能碳基催化劑。
通過對制得的鈷鉬復合金屬多功能碳基催化劑進行表征,表征結果如圖1、圖2所示。
圖1為鈷鉬復合金屬多功能碳基催化劑的電鏡掃描圖(SEM),顯示了鈷鉬碳化物具有20-50nm有規則的微孔。
圖2為鈷鉬復合金屬多功能碳基催化劑的透射電鏡圖(TEM)圖像,鈷鉬碳化物微孔主要為20-40nm,TEM圖像表明碳熱還原過程中,碳作為還原劑參與反應,增加了微孔數量;而氫作為還原劑可使鈷鉬碳結構更加牢固,碳用量也相對減少。
實施例2。取3g實施例1中步驟d制得的催化劑加入0.5英寸不銹鋼反應器中,先通30min氦氣將系統排空,然后通入N2-H2(1:1)的混合氣體在400℃下保溫8小時進行預還原反應,隨后考察催化劑在溫度分別為350℃、380℃、410℃下的催化活性。表1是CO轉化率、C1-C4輕質烴和液態產品選擇性隨溫度變化的結果。其反應條件:壓力1000psi,合成氣50%H2+50%CO(模擬甲烷干重整制備所得的合成氣),反應空速(GHSV)3000h-1,運行時間8~20小時。由表1可知CO轉化率隨溫度的增加而增加。
表1 CO轉化率、C1-C4輕質烴和液態產品選擇性隨溫度的變化反應條件
實施例3。取3g實施例1中步驟d制得的催化劑按實施例2進行預還原反應,隨后考察催化劑在壓力200psi、500psi、800psi的催化活性。表2是CO轉化率、C1-C4輕質烴和液態產品選擇性隨壓力的變化的結果,其反應條件:溫度380℃,合成氣50%H2+50%CO,反應空速(GHSV)3000h-1,運行時間48~100小時。由表2可知CO轉化率隨壓力的增加而增加。
表2 CO轉化率、C1-C4輕質烴和液態產品選擇性隨壓力的變化反應條件
實施例4。取3g實施例1中步驟d制得的催化劑按實施例2進行預還原反應,隨后考察催化劑在反應空速1000h-1、3000h-1、5000h-1的催化活性。表3是CO轉化率、C1-C4輕質烴和液態產品選擇性隨反應空速的變化的結果。其反應條件:溫度380℃,壓力1000psi,合成氣50.0%H2+50.0%CO。由表3可知CO轉化率隨反應空速的增加而降低。
表3 CO轉化率、C1-C4輕質烴和液態產品選擇性隨反應空速的變化反應條件
實施例5。取3g實施例1中步驟d制得的催化劑按實施例2進行預還原反應,隨后考察催化劑在合成氣組分50.0%H2+50.0%CO,67.0%H2+33.0%CO,19.0%H2+20.0%CO+12.0%CO2+1.921%CH4+N2(生物質衍生合成氣)的催化活性。表4是CO轉化率、C1-C4輕質烴和液態產品選擇性隨合成氣組分的變化的結果。其反應條件:溫度380℃,壓力1000psi,反應空速(GHSV)3000h-1。由表4可知該催化劑在富氮的合成氣合成烴時具有較高的活性和選擇性。
表4 CO轉化率、C1-C4輕質烴和液態產品選擇性隨合成氣組分的變化反應條件
實施例6。取3g實施例1中步驟d制得的催化劑按實施例2進行預還原反應,隨后考察催化劑在運行時間(0~2000小時)的催化活性。圖3是CO轉化率、C1-C4輕質烴和液態產品選擇性隨運行時間的變化的結果。其反應條件:溫度380℃,壓力1000psi,反應空速3000h-1,合成氣50.0%H2+50.0%CO。圖3中催化劑在工況下運行40小時后CO轉化率達到了81%,并在運行2000小時后CO轉化率仍保持不變,可知反應中催化劑非常穩定。
實施例7。取3g實施例1中步驟b制得的催化劑按實施例2進行預還原反應,隨后考察實施例1中步驟b與步驟d催化劑的催化活性。表5是實施例1中步驟b中CO轉化率、C1-C4輕質烴和液態產品選擇性的結果,其反應條件:溫度380℃,壓力1000psi,反應空速3000h-1,合成氣組分50.0%H2+50.0%CO。
表5實施例1中步驟b催化劑的CO轉化率、C1-C4輕質烴和液態產品選擇性
實施例8。同實施例1,只是步驟c中添加的過渡金屬為銠、鉑和釕,添加量為1.0wt%,再取3g實施例1步驟d制備的催化劑,按實施例2進行預還原反應,隨后考察催化劑在添加1.0wt%Rh/Pt/Ru后的催化活性。表6是Rh/Pt/Ru對催化劑活性的影響,表7~10是含1.0wt%銠的催化產物中酯、醛、酮和其他含氧化合物的典型液相產物分布。其反應條件:合成氣50.0%H2+50.0%CO,溫度380℃,壓力1000psi,反應空速3000h-1。表6中附加過渡金屬催化劑對產物中CO轉化率及烴類選擇性都有不同程度的改善。
表6 Rh/Pt/Ru對催化劑活性的影響
表7含銠催化劑催化產物中酯的典型產物分布
表8含銠催化劑催化產物中醛和酮的典型產物分布
表9含銠催化劑催化產物中醇的典型產物分布
表10含銠催化劑催化產物中含氧化合物的典型產物分布
實施例9。同實施例1,只是步驟c中添加的過渡金屬為釩、鎳和鎢,添加量為20wt%,再取3g實施例1步驟d制備的催化劑,按實施例2進行預還原反應,隨后考察催化劑在添加20wt%V/Ni/W后的催化活性。表11是V/Ni/W對催化劑活性的影響,表11中附加過渡金屬催化劑對產物中CO轉化率及烴類選擇性都有不同程度的改善。
表11 V/Ni/W對催化劑活性的影響
實施例10。同實施例1,只是步驟c中添加的過渡金屬為鐵,添加量為10.0wt%,再取3g實施例1步驟d制備的催化劑,按實施例2進行預還原反應,隨后考察催化劑在添加10.0wt%Fe后的催化活性。表12是不同溫度下費托合成中CO轉化率,CO2和烴類選擇性。其反應條件:壓力1000psi,反應空速2000h-1,生物質衍生合成氣47%N2+21%CO+18%H2+12%CO2+2%CH4,運行時間48小時。
表12不同溫度下費托合成中CO轉化率,CO2和烴類選擇性
表12結果表明溫度升高時CO轉化率增加,在350℃時可達到98.3%。液態產物主要有烴類,包括鏈烷烴,異鏈烷烴,芳烴,環烷烴(環烷)和烯烴在內的汽油、航空燃料、煤油和柴油;除了烴之外,液態油相中也含有5.0%的含氧化合物,分別是異丙醇,乙醇,叔丁醇和甲醇。
實施例11。一種鈷鉬復合金屬多功能碳基催化劑的制備方法,制備過程包括以下步驟:
a、用五氯化鉬和氯化鈷配制鈷鉬摩爾比為1:1的鹽溶液,步驟如下:11.47g五氯化鉬溶于20mL去離子水中,將該溶液加熱至80℃,攪拌至澄清液,然后再將溶液冷卻至40℃并加入9.99g氯化鈷,再次攪拌至澄清液,隨后把制得的雙金屬鹽溶液逐滴加入盛有10g炭黑的燒杯中并不斷攪拌,并在80℃下蒸發去除溶劑,剩余固體在110℃下干燥12小時,得A品;
b、將步驟a中所得固體A品送入外徑1英寸的石英反應管,采用程序控溫加熱管式爐,樣品量約為20~200g/次,在500mL/min氦氣流中加熱至300℃,保溫2小時,然后在100~1000mL/min氦氣流或純氫氣流中以1~100℃/min的速率加熱至700~1100℃,保溫2小時,最后樣品在室溫下通入含微量氧氣的氮氣2小時進行鈍化處理,得B品;
c、取0.5gCuO溶于10mL去離子水,將CuO溶液逐滴加入步驟b所得的B品中并不斷攪拌,并在80℃下蒸發去除溶劑,得到的樣品在110℃下干燥12小時,得C品;
d、取0.5gNaNO3溶于10mL去離子水,將NaNO3溶液逐滴加入步驟c所得C品中并不斷攪拌,并在80℃下蒸發去除溶劑,得到的樣品在110℃下干燥12小時,最后將樣品置于350℃氮氣流中焙燒3小時,粉碎造粒即得鈷鉬復合金屬多功能碳基催化劑。
實施例12。一種鈷鉬復合金屬多功能碳基催化劑的制備方法,制備過程包括以下步驟:
a、用六羰基鉬和乙酸鈷配制鈷鉬摩爾比為1:1的鹽溶液,步驟如下:11.09g六羰基鉬溶于20mL去離子水中,將該溶液加熱至80℃,攪拌至澄清液,然后再將溶液冷卻至40℃并加入4.96g乙酸鈷,再次攪拌至澄清液,隨后把制得的雙金屬鹽溶液逐滴加入盛有10g碳納米管的燒杯中并不斷攪拌,并在80℃下蒸發去除溶劑,剩余固體在110℃下干燥12小時,得A品;
b、將步驟a中所得固體A品送入外徑1英寸的石英反應管,采用程序控溫加熱管式爐,樣品量約為20~200g/次,在500mL/min氦氣流中加熱至300℃,保溫2小時,然后在100~1000mL/min氦氣流或純氫氣流中以1~100℃/min的速率加熱至700~1100℃,保溫2小時,最后樣品在室溫下通入含微量氧氣的氮氣2小時進行鈍化處理,得B品;
c、取0.5g AgNO3溶于10mL去離子水,將AgNO3溶液逐滴加入步驟b所得的B品中并不斷攪拌,并在80℃下蒸發去除溶劑,得到的樣品在110℃下干燥12小時,得C品;
d、取0.5g KCl溶于10mL去離子水,將KCl溶液逐滴加入步驟c所得C品中并不斷攪拌,并在80℃下蒸發去除溶劑,得到的樣品在110℃下干燥12小時,最后將樣品置于350℃氮氣流中焙燒3小時,粉碎造粒即得鈷鉬復合金屬多功能碳基催化劑。
實施例13。一種鈷鉬復合金屬多功能碳基催化劑的制備方法,制備過程包括以下步驟:
a、用四氯氧化鉬和乙酰丙酮鈷配制鈷鉬摩爾比為1:1的鹽溶液,步驟如下:10.66g四氯氧化鉬溶于20mL去離子水中,將該溶液加熱至80℃,攪拌至澄清液,然后再將溶液冷卻至40℃并加入10.80g乙酰丙酮鈷,再次攪拌至澄清液,隨后把制得的雙金屬鹽溶液逐滴加入盛有10g碳納米纖維的燒杯中并不斷攪拌,并在80℃下蒸發去除溶劑,剩余固體在110℃下干燥12小時,得A品;
b、將步驟a中所得固體A品送入外徑1英寸的石英反應管,采用程序控溫加熱管式爐,樣品量約為20~200g/次,在500mL/min氦氣流中加熱至300℃,保溫2小時,然后在100~1000mL/min氦氣流或純氫氣流中以1~100℃/min的速率加熱至700~1100℃,保溫2小時,最后樣品在室溫下通入含微量氧氣的氮氣2小時進行鈍化處理,得B品;
c、取0.5g PdCl2溶于10mL去離子水,將PdCl2溶液逐滴加入步驟b所得的B品中并不斷攪拌,并在80℃下蒸發去除溶劑,得到的樣品在110℃下干燥12小時,得C品;
d、取0.5g KOH溶于10mL去離子水,將KOH溶液逐滴加入步驟c所得C品中并不斷攪拌,并在80℃下蒸發去除溶劑,得到的樣品在110℃下干燥12小時,最后將樣品置于350℃氮氣流中焙燒3小時,粉碎造粒即得鈷鉬復合金屬多功能碳基催化劑。
實施例14。一種鈷鉬復合金屬多功能碳基催化劑的制備方法,制備過程包括以下步驟:
a、用三氧化鉬和七水合硫酸鈷配制鈷鉬摩爾比為1:1的鹽溶液,步驟如下:6.05g三氧化鉬溶于20mL去離子水中,將該溶液加熱至80℃,攪拌至澄清液,然后再將溶液冷卻至40℃并加入11.81g七水合硫酸鈷,再次攪拌至澄清液,隨后把制得的雙金屬鹽溶液逐滴加入盛有10g石墨烯的燒杯中并不斷攪拌,并在80℃下蒸發去除溶劑,剩余固體在110℃下干燥12小時,得A品;
b、將步驟a中所得固體A品送入外徑1英寸的石英反應管,采用程序控溫加熱管式爐,樣品量約為20~200g/次,在500mL/min氦氣流中加熱至300℃,保溫2小時,然后在100~1000mL/min氦氣流或純氫氣流中以1~100℃/min的速率加熱至700~1100℃,保溫2小時,最后樣品在室溫下通入含微量氧氣的氮氣2小時進行鈍化處理,得B品;
c、取0.5g PdCl2溶于10mL去離子水,將PdCl2溶液逐滴加入步驟b所得的B品中并不斷攪拌,并在80℃下蒸發去除溶劑,得到的樣品在110℃下干燥12小時,得C品;
d、取0.5g KOH溶于10mL去離子水,將KOH溶液逐滴加入步驟c所得C品中并不斷攪拌,并在80℃下蒸發去除溶劑,得到的樣品在110℃下干燥12小時,最后將樣品置于350℃氮氣流中焙燒3小時,粉碎造粒即得鈷鉬復合金屬多功能碳基催化劑。
實施例15。一種鈷鉬復合金屬多功能碳基催化劑的制備方法,制備過程包括以下步驟:
a、用乙酸鉬和四氧化三鈷配制鈷鉬摩爾比為1:1的鹽溶液,步驟如下:8.99g乙酸鉬溶于20mL去離子水中,將該溶液加熱至80℃,攪拌至澄清液,然后再將溶液冷卻至40℃并加入3.34g四氧化三鈷,再次攪拌至澄清液,隨后把制得的雙金屬鹽溶液逐滴加入盛有10g生物炭的燒杯中并不斷攪拌,并在80℃下蒸發去除溶劑,剩余固體在110℃下干燥12小時,得A品;
b、將步驟a中所得固體A品送入外徑1英寸的石英反應管,采用程序控溫加熱管式爐,樣品量約為20~200g/次,在500mL/min氦氣流中加熱至300℃,保溫2小時,然后在100~1000mL/min氦氣流或純氫氣流中以1~100℃/min的速率加熱至700~1100℃,保溫2小時,最后樣品在室溫下通入含微量氧氣的氮氣2小時進行鈍化處理,得B品;
c、取0.5g PdCl2溶于10mL去離子水,將PdCl2溶液逐滴加入步驟b所得的B品中并不斷攪拌,并在80℃下蒸發去除溶劑,得到的樣品在110℃下干燥12小時,得C品;
d、取0.5g KOH溶于10mL去離子水,將KOH溶液逐滴加入步驟c所得C品中并不斷攪拌,并在80℃下蒸發去除溶劑,得到的樣品在110℃下干燥12小時,最后將樣品置于350℃氮氣流中焙燒3小時,粉碎造粒即得鈷鉬復合金屬多功能碳基催化劑。
以上所述是本發明的優選實施方式而已,當然不能以此來限定本發明之權利范圍,應當指出,對于本技術領域的普通技術人員來說,在不脫離本發明原理的前提下,還可以做出若干改進和變動,這些改進和變動也視為本發明的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!