一種貴金屬微區鑲嵌型氧化物復合材料的制備方法與流程
本發明涉及無機復合材料,尤其是涉及一種貴金屬微區鑲嵌型氧化物復合材料的制備方法。
背景技術:
貴金屬納米顆粒作為一類近年來被廣泛研究的新型分散態材料,已經在生物、催化、電子技術以及分析測試技術等領域逐步得到具體而切實的應用。基于納米材料的量子尺寸效應,當金屬顆粒小于一定維度時,費米能級附近的電子能級將由連續態轉化為離散態,并因此賦予了納米金屬粒子獨特的電學和化學反應特性。在以原子級別的相互作用為基礎的化學鍵合方式重組的場合下,金屬能級狀態的改變使其參與化學反應的行為發生了重大的調整,尤其在具有反應累加特征的催化作用過程中,該效應表現的更為明顯。
由于受到納米粒子本身具有的高表面能所驅動,在顆粒接觸的條件下,將趨向粒子團聚并融合,同時也將消解尺寸效應所帶來的反應特性的改變,所以通常采用保護劑或擔載的方式抑制顆粒間的接觸,以維持顆粒間的分散。目前采用的分散技術主要有包覆型保護劑法與高比表面載體擔載法兩種模式,保護劑法利用吸附在納米顆粒表面的有機基團(樹狀分子鏈或高級脂肪酸鏈)形成的空間位阻防止顆粒間相互接觸并融合,而高比表面載體則提供了納米顆粒較大的二維分布空間,從而降低顆粒間相互接近的幾率,實現高分散。保護劑分子在納米粒子表面的吸附在抑制團聚的同時也阻塞了金屬粒子的反應位置,這就要求納米金屬粒子進入工作狀態前須對保護劑予以消除,暴露活性表面,而保護劑脫除后的納米粒子分散性一般仍要采用擔載的方式進行維持,因此如活性炭、氧化鋁、氧化鋯、硅膠以及分子篩等各類高比表面材料在納米金屬催化劑的設計中通常是不可或缺的重要組成。
落位在載體上的納米金屬粒子失去配體之后,趨向于從周圍環境中尋找成鍵機會,緩解低配位并降低表面能,由于各顆粒已經散落至廣闊的載體表面上,因此再次形成鍵合一般只能通過氣相氧化與羥基化的形式或與載體共享陰離子表層(如碳載體表面的含氧官能團以及氧化物表面的氧離子或氫氧根離子)。受到較高的粒子表面能與脫除保護劑所經歷的高溫處理過程的影響,金屬顆粒與載體表面易于發生程度較強的相互作用(SMI),進而對金屬組分微晶的存在形態形成重要的調控效應,另外,由于氧化物載體的晶型、缺陷以及離子遷移性等諸多結構特征在表面的延伸,也會通過上述SMI的作用機制,在塑造負載型納米金屬微晶的最終形態上發揮重要影響。
復合材料的性能表現與其單元組分的相對分布方式密切相關,而制備路線中各組分的引入方式則決定了金屬與氧化物擔體的相互作用形式。負載型催化材料通常采用浸漬法、共沉淀或離子交換法將金屬組分引入到氧化物載體表面[1-3],而具有疏水特征的納米金屬粒子的保護劑卻難以對氧化物極性表面形成有效浸潤而使得相應載體常限于碳、碳化硅與氧化硅等疏水性載體[4],因此上述幾類方法均不易實現兩種單元組分的均勻混合,從而導致擔載型金屬/氧化物體系易發生如下兩種劣質化效應:一是納米金屬的疏水性分散介質在氧化物載體粉末聚集體的外表面因無法克服表面張力的能壘而凝聚成相,隨著溶劑的移除最終造成金屬組分過度富集;另外,如采用金屬離子/極性溶劑形式引入氧化物載體表面,則造成金屬組分優先以離子形式與載體組分發生強相互作用,從而喪失其金屬聚集態的反應特性。
技術實現要素:
本發明的目的在于通過對凝膠型氧化物載體的前體進行表面非極性化改造,使其在非極性溶劑中與納米金屬粒子分散系形成浸潤,從而將具有表面疏水性保護劑包覆的貴金屬納米顆粒引入非極性化表面,在控制金屬/載體相互作用程度的前提下,實現貴金屬組分以金屬聚集態的形式分散在極性氧化物載體表面,以維持其反應特性的一種貴金屬微區鑲嵌型氧化物復合材料的制備方法。
本發明包括以下步驟:
1)配制金屬氧化物載體的組分陽離子鋯、鋁的水溶液,或金屬氧化物載體的組分陽離子鋯、鋁的水溶液與其它堿土或稀土修飾離子的混合金屬鹽的水溶液,調節溶液pH值以形成凝膠;分去水層,以乙酰丙酮對凝膠中的羥基進行置換并分去置換反應生成的水,反應后獲得表面乙酰丙酮化的氫氧化物凝膠載體;
2)在氮氣保護下,將步驟1)中所獲得的表面乙酰丙酮化的氫氧化物凝膠載體轉移至正辛醚溶劑中,將現場或離線合成的單組分貴金屬或貴金屬合金納米顆粒引入,升溫反應,直至納米顆粒在載體表面落位完成,降溫并沉降后,分去溶劑層,得懸浮液;
3)將步驟2)得到的懸浮液沉降后分去辛醚溶劑,煅燒除去復合材料中的各類有機助劑,再將煅燒產物還原,制得貴金屬微區鑲嵌型氧化物復合材料。
在步驟1)中,所述金屬氧化物載體的組分陽離子鋯、鋁的水溶液,所使用的金屬鹽可選自鋯的氯化物、鋁的氯化物、硝酸鹽、硫酸鹽等中的一種;所述其它堿土或稀土修飾離子可采用堿土金屬元素鎂、鈣、鍶、鋇與稀土金屬元素鑭、鈰、鐠、釹、釤、銪、釓、鋱的硝酸鹽的形式引入至混合離子溶液中;所述調節溶液pH值可采用氨水、碳酸銨、碳酸氫銨等中至少一種化合物復配形成的堿性水溶液;所述分去水層的方法可采用傾析法或抽濾法;所述以乙酰丙酮對凝膠中的羥基進行置換并分去置換反應生成的水的方法可在攪拌并加熱下進行,置換反應產生的水通過減壓蒸餾或流動氣體帶出。
在步驟2)中,所述現場合成單組分貴金屬納米顆粒可通過將釕、銠、鈀、銥、鉑的氯化物、乙酸鹽、乙酰丙酮鹽在乙酰丙酮中溶解后,加入合成體系中,并升溫至120℃,在120℃加熱攪拌下滴入α二醇(乙二醇、1,2-丙二醇或1,2-十二烷二醇)與油胺的辛醚溶液,滴加完畢后,升溫至140℃蒸出乙酰丙酮溶劑后,維持反應30min,可繼續升溫至200℃或300℃。如使用離線合成的單組分貴金屬或貴金屬合金納米顆粒,可將其分散至辛醚體系后,直接升溫蒸出溶劑,并在200℃或300℃恒溫至反應完成。
在步驟3)中,所述沉降后分去辛醚溶劑可采用離心或抽濾的方法;所述煅燒的氣氛為氧化性氣氛,所述氧化性氣氛可選擇空氣至按體積百分比純氧為20%~100%之間濃度范圍內的氧化性氣體;所述煅燒的溫度可為450~750℃;所述煅燒產物還原是將復合材料體系中的貴金屬組分轉化為單質狀態,可選擇氫氣、一氧化碳、烴及其相應的稀釋氣體,還原溫度一般低于煅燒溫度100~200℃,優選300~550℃。
本發明的載體為氧化鋁、氧化鋯以及上述載體經稀土或堿土改造而形成的復合氧化物載體,該氧化物載體在制備前體階段,能夠形成氫氧化物凝膠;所述納米金屬顆粒為現場或離線合成的單組分貴金屬或貴金屬合金的納米顆粒,金屬粒子由疏水性保護劑分子配位,能夠在非極性溶劑中形成穩定分散系。
本發明基于貴金屬納米粒子分散系的特點,對載體前體進行非極性化改造,在維持金屬粒子聚集態的前提下,突破非極性溶劑在氧化物型載體表面的分散阻力,從而在抑制金屬/載體強相互作用程度的前提下,實現納米金屬粒子在復合材料中的均勻分布并維持其反應特性。
本發明采用氫氧化物凝膠型前體為原料,經過表面部分乙酰丙酮化改造,形成非極性表面,與非極性溶劑中所含有的表面保護劑修飾的貴金屬納米金屬微粒在近軟化點進行相容性負載,繼續通過加熱氧化移除有機組分(表面改性劑與保護劑分子),在氧化物型載體與納米顆粒的SMI作用下自組裝為貴金屬微區鑲嵌型復合材料。在復合材料中,因基體織構形態已經由氫氧化物前體確定,貴金屬對其形成的修飾效果能夠實現在表面的均勻分布,未對其內部組成造成影響,基體的結構穩定性可得到保障;另外貴金屬組分分布在氧化物基體的外層,也減少了內部包夾對貴金屬成分的浪費。此外,貴金屬聚集與其在基體材料上的分散形態是其化學與電子特性的物質結構基礎,因此,在材料表層嵌入還原形態的貴金屬微區,并通過載體對金屬微區在原子水平的加工效應,能夠實現對復合材料吸附、催化、分離、光學等實用功能的改善性調變。
附圖說明
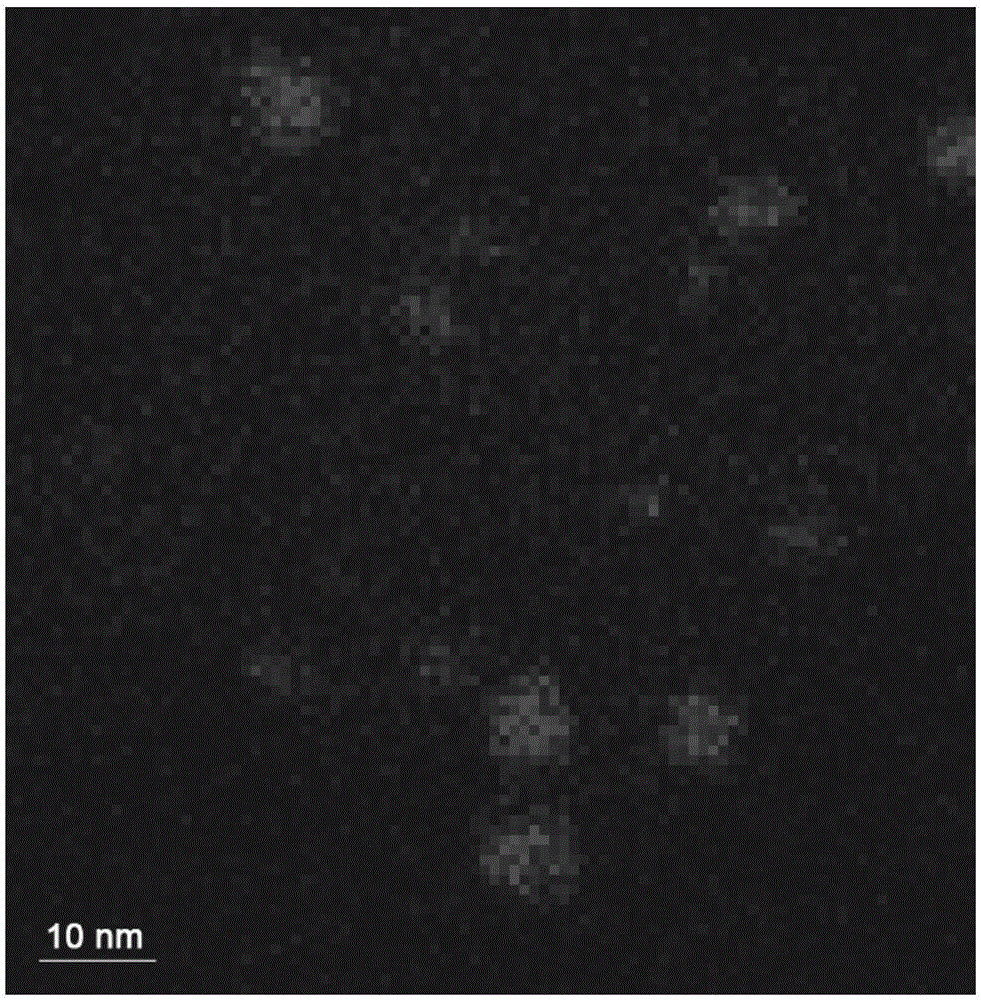
圖1為鈀金屬微區結構修飾氧化鋯復合材料的HAADF-STEM圖像。
圖2為選區中鈀組分的元素分布形態(面掃)。
具體實施方式
實施例1
在含有1.5g氯化鎂與40g硫酸鋁的混鹽離子溶液中滴入氨水與碳酸氫銨的混合沉淀劑,將pH調節至8~9;采用傾析法分去水層,經洗滌去除氯離子;將上述制得的混合離子堿式碳酸鹽凝膠加入至250ml乙酰丙酮中,室溫下激烈攪拌20h;加入300mL正辛醚并在攪拌下蒸出120℃的前餾分,維持20mmHg真空度,在90℃繼續反應1h。
在強烈攪拌下,于上述混合金屬離子堿式碳酸鹽凝膠懸浮液中,加入含有0.2g乙酰丙酮鈀的乙酰丙酮溶液;攪拌均勻后,逐滴加入含有1.8g的1,2-十二烷二醇與0.93g的油胺的辛醚溶液(滴加速度15d/min);滴加完畢后,升溫蒸出140℃前餾分,并維持攪拌反應30min;繼續升溫至260℃,持續回流2h,冷卻。
上述獲得的懸浮液經沉降并離心分離出固形物,轉移至剛玉舟中;在500℃的管式爐中通入空氣進行煅燒30min;降溫后,提高氧氣比例至50vol.%升溫至500℃,繼續煅燒30min,降溫;煅燒氣氛切換為5%H2(Ar平衡氣),升溫至400℃,還原2h;冷卻,獲得產品。
實施例2
在含有25g硝酸氧鋯與3.1g硝酸鈰及1.5g硝酸鋱的混鹽離子溶液中滴入氨水,將pH調節至9~10;采用抽濾法分去水層,濾餅經水洗滌去除硝酸根;將上述制得的混合離子氫氧化物凝膠加入盛有250ml乙酰丙酮的敞口體系中,50℃下激烈攪拌30h;加入300mL正辛醚并在攪拌下蒸出120℃的前餾分,在120℃繼續反應1h。
在強烈攪拌下,于上述混合金屬離子氫氧化物凝膠懸浮液中,加入含有0.8g乙酰丙酮鉑的乙酰丙酮溶液;攪拌均勻后,逐滴加入含有1.8g的1,2-十二烷二醇與0.4g油酸及0.93g的油胺的辛醚溶液(滴加速度15d/min);滴加完畢后,升溫蒸出140℃前餾分,并維持攪拌反應60min;繼續升溫至280℃,持續回流1.5h,冷卻。
上述獲得的懸浮液經沉降并離心分離出固形物,轉移至剛玉舟中;在500℃的管式爐中通入空氣進行煅燒60min,降溫;焙燒氣氛切換為H2,升溫至300℃,還原2h;冷卻,獲得產品。
實施例3
在含有25g硝酸氧鋯與0.95g硝酸鍶的混鹽離子溶液中滴入氨水與碳酸氫銨的混合沉淀劑,將pH調節至8;采用抽濾法分去水層,濾餅經水洗滌去除硝酸根;將上述制得的混合離子堿式碳酸鹽凝膠加入至200ml乙酰丙酮中,室溫下激烈攪拌20h;加入300mL苯甲醚并在攪拌下蒸出120℃的前餾分,在120℃繼續反應1h。
在強烈攪拌下,于上述混合金屬離子堿式碳酸鹽凝膠懸浮液中,加入含有0.6g乙酰丙酮鈀的乙酰丙酮溶液;攪拌均勻后,逐滴加入含有1.8g的1,2-十二烷二醇與0.93g的油胺的苯甲醚溶液(滴加速度15d/min);滴加完畢后,升溫蒸出140℃前餾分,并維持攪拌反應30min;繼續升溫至260℃,持續回流2h,冷卻。
上述獲得的懸浮液經沉降并離心分離出固形物,轉移至剛玉舟中;在750℃的管式爐中通入空氣進行煅燒60min,降至室溫;氮氣吹掃后,將煅燒氣氛切換為5%H2(Ar平衡氣),升溫至550℃,還原2h;冷卻,獲得產品。
實施例4
在含有73g硝酸鋁與1.2g硝酸鑭的混鹽離子溶液中滴入氨水與碳酸氫銨的混合沉淀劑,將pH調節至9;采用傾析法分去水層,經水洗滌去除氯離子后;將上述制得的混合離子堿式碳酸鹽凝膠加入至200ml乙酰丙酮中,室溫下激烈攪拌20h;加入300mL正辛醚并在攪拌下蒸出120℃的前餾分,在120℃繼續反應1h。
在強烈攪拌下,于上述混合金屬離子堿式碳酸鹽凝膠懸浮液中,加入含有0.15g乙酰丙酮鈀的乙酰丙酮溶液;攪拌均勻后,逐滴加入含有1.8g的1,2-十二烷二醇與0.93g的油胺的辛醚溶液(滴加速度15d/min);滴加完畢后,升溫蒸出140℃前餾分,并維持攪拌反應30min;繼續升溫至280℃,持續回流2h,冷卻。
上述獲得的懸浮液經沉降并離心分離出固形物,轉移至剛玉舟中;在500℃的管式爐中通入空氣進行煅燒30min;降溫后,提高氧氣比例至50vol.%升溫至500℃,繼續煅燒30min,降溫;煅燒氣氛切換為氫氣,升溫至400℃,還原2h;冷卻,獲得產品。
實施例5
在含有25g硝酸氧鋯與3.1g硝酸釤的混鹽離子溶液中滴入氨水與碳酸氫銨的混合沉淀劑,將pH調節至8~9;采用傾析法分去水層,經水洗滌去除硝酸根離子后;將上述制得的混合離子堿式碳酸鹽凝膠加入至200ml乙酰丙酮中,室溫下激烈攪拌20h;加入300mL正辛醚并在攪拌下蒸出120℃的前餾分,維持20mmHg真空度,在90℃繼續反應1h。
在強烈攪拌下,于上述混合金屬離子堿式碳酸鹽凝膠懸浮液中,加入含有0.5g乙酰丙酮鉑的乙酰丙酮溶液;攪拌均勻后,逐滴加入含有0.5g的乙二醇與0.6g油酸及0.93g的油胺的辛醚溶液(滴加速度15d/min);滴加完畢后,升溫蒸出140℃前餾分,并維持攪拌反應30min;繼續升溫至260℃,持續回流2h,冷卻。
上述獲得的懸浮液經沉降并離心分離出固形物,轉移至剛玉舟中;在500℃的管式爐中通入空氣進行煅燒30min;降溫后,提高氧氣比例至50vol.%升溫至500℃,繼續煅燒30min,降溫;將煅燒氣氛切換為10%H2(Ar平衡氣),升溫至450℃,還原2h;冷卻,獲得產品。
實施例6
在含有25g硝酸氧鋯溶液中滴入氨水沉淀劑,將pH調節至10;采用抽濾法除去水層,經水洗滌去除硝酸根離子后;將上述制得的氫氧化鋯凝膠加入至200ml乙酰丙酮中,80℃下激烈攪拌20h;加入300mL正辛醚并在攪拌下蒸出120℃的前餾分,維持20mmHg真空度,在90℃繼續反應1h。
在強烈攪拌下,于上述凝膠懸浮液中,加入含有0.1g納米金屬鈀顆粒的正辛醚溶液(滴加速度15d/min);滴加完畢后,升溫蒸出140℃前餾分,并維持攪拌反應30min,冷卻。
上述獲得的懸浮液經沉降并離心分離出固形物,轉移至剛玉舟中;在500℃的管式爐中通入空氣進行煅燒30min;降溫后,提高氧氣比例至50vol.%升溫至500℃,繼續煅燒30min,降溫;將煅燒氣氛切換為10%H2(Ar平衡氣),升溫至450℃,還原2h;冷卻,獲得產品。
實施例7
在40g硝酸鋯與2.0g硝酸鈣的混鹽離子溶液中滴入氨水與碳酸氫銨的混合沉淀劑,將pH調節至8~9;采用傾析法分去水層,經水洗滌去除硝酸根離子后;將上述制得的混合離子堿式碳酸鹽凝膠加入至200ml乙酰丙酮中,室溫下激烈攪拌20h;加入300mL正辛醚并在攪拌下蒸出120℃的前餾分,在120℃繼續反應1h。
在強烈攪拌下,于上述混合金屬離子堿式碳酸鹽凝膠懸浮液中,加入含有0.1g乙酰丙酮鉑的乙酰丙酮溶液;攪拌均勻后,逐滴加入含有1g的1,2十二烷二醇與0.9g的油胺的辛醚溶液(滴加速度15d/min);滴加完畢后,升溫蒸出140℃前餾分,并維持攪拌反應30min;繼續升溫至280℃,持續回流2h,冷卻。
上述獲得的懸浮液經沉降并離心分離出固形物,轉移至剛玉舟中;在700℃的管式爐中通入空氣進行煅燒30min;降溫后,提高氧氣比例至50vol.%升溫至500℃,繼續煅燒30min,降溫;將煅燒氣氛切換為10%H2(Ar平衡氣),升溫至450℃,還原2h;冷卻,獲得產品。
實施例8
在21g氯化氧鋯與1.0g硝酸釤的混鹽離子溶液中滴入氨水與碳酸氫銨的混合沉淀劑,將pH調節至8;采用傾析法分去水層,經水洗滌去除氯離子后;將上述制得的混合離子堿式碳酸鹽凝膠加入至200ml乙酰丙酮中,室溫下激烈攪拌20h;加入300mL正辛醚并在攪拌下蒸出120℃的前餾分,在120℃繼續反應1h。
在強烈攪拌下,于上述混合金屬離子堿式碳酸鹽凝膠懸浮液中,加入含有0.15g乙酰丙酮鈀的乙酰丙酮溶液;攪拌均勻后,逐滴加入含有1.8g的1,2十二烷二醇與0.93g的油胺的辛醚溶液;滴加完畢后,升溫蒸出140℃前餾分,并維持攪拌反應30min;繼續升溫至260℃,持續回流2h,冷卻。
上述獲得的懸浮液經沉降并離心分離出固形物,轉移至剛玉舟中;在500℃的管式爐中通入空氣進行煅燒30min;降溫后,提高氧氣比例至50vol.%升溫至500℃,繼續煅燒30min,降溫;將煅燒氣氛切換為5%H2(Ar平衡氣),升溫至400℃,還原2h;冷卻,獲得產品。
實施例9
在73g硝酸鋁與1.5g硝酸釹鐠的混鹽離子溶液中滴入氨水與碳酸氫銨的混合沉淀劑,將pH調節至9;采用傾析法分去水層,經水洗滌去除硝酸根離子后;將上述制得的混合離子堿式碳酸鹽凝膠加入至200ml乙酰丙酮中,室溫下激烈攪拌20h;加入300mL正辛醚并在攪拌下蒸出120℃的前餾分,維持20mmHg真空度,在90℃繼續反應1h。
在強烈攪拌下,于上述凝膠懸浮液中,加入含有納米金屬鉑顆粒的辛醚溶液;滴加完畢后,升溫蒸出140℃前餾分,并維持攪拌反應30min,冷卻。
上述獲得的懸浮液經沉降并離心分離出固形物,轉移至剛玉舟中;在500℃的管式爐中通入空氣進行煅燒60min,降溫;將煅燒氣氛切換為5%H2(Ar平衡氣),升溫至400℃,還原2h;冷卻,獲得產品。
實施例10
在25g硝酸氧鋯與0.5g乙酸銪的混鹽離子溶液中滴入氨水與碳酸氫銨的混合沉淀劑,將pH調節至9;采用傾析法分去水層,經水洗滌去除硝酸根離子后;將上述制得的混合離子堿式碳酸鹽凝膠加入至200ml乙酰丙酮中,室溫下激烈攪拌20h;加入300mL正辛醚并在攪拌下蒸出120℃的前餾分,維持20mmHg真空度,在90℃繼續反應1h。
在強烈攪拌下,于上述凝膠懸浮液中,加入含有0.1g納米金屬鈀顆粒的辛醚溶液;滴加完畢后,升溫蒸出140℃前餾分,并維持攪拌反應30min,冷卻。
上述獲得的懸浮液經沉降并離心分離出固形物,轉移至剛玉舟中;在500℃的管式爐中通入空氣進行煅燒60min,降溫;將煅燒氣氛切換為5%H2(Ar平衡氣),升溫至400℃,還原2h;冷卻,獲得產品。
實施例11
在25g硝酸鋯與0.8g氧化釓反應形成的澄清離子混鹽溶液中滴入氨水與碳酸氫銨的混合沉淀劑,將pH調節至8;采用傾析法分去水層,經水洗滌去除硝酸根離子后;將上述制得的混合離子堿式碳酸鹽凝膠加入至200ml乙酰丙酮中,室溫下激烈攪拌20h;加入300mL正辛醚并在攪拌下蒸出120℃的前餾分,維持20mmHg真空度,在90℃繼續反應1h。
在強烈攪拌下,于上述凝膠懸浮液中,加入含有0.1g納米金屬鈀顆粒的辛醚溶液;滴加完畢后,升溫蒸出140℃前餾分,并維持攪拌反應30min,冷卻。
上述獲得的懸浮液經沉降并離心分離出固形物,轉移至剛玉舟中;在500℃的管式爐中通入空氣進行煅燒60min,降溫;將煅燒氣氛切換為5%H2(Ar平衡氣),升溫至400℃,還原2h;冷卻,獲得產品。
鈀金屬微區結構修飾氧化鋯復合材料的HAADF-STEM圖像參見圖1,選區中鈀組分的元素分布形態(面掃)參見圖2。
- 還沒有人留言評論。精彩留言會獲得點贊!