一種CO脫氫凈化用Pd催化劑的原位再生方法與流程
本發明涉及一種失活后的Pd催化劑的原位再生方法,應用于工業CO氣體脫氫凈化工藝。
背景技術:
在工業用催化劑的運行過程中,不可避免的會出現隨著反應時間的延長,催化劑的活性以及選擇性逐漸降低,催化劑的效率不斷降低并最終失活的現象。催化劑失活的主要類型分為積炭失活、中毒失活和燒結失活。通常,在工業生產過程中為保證反應的穩定運行,必須要定期更換催化劑。由于貴金屬資源稀缺,價格相對昂貴,對于貴金屬催化劑來說,催化劑的再生可以實現貴金屬的重復利用,減少催化劑更換頻率,極大節省了催化劑的運營成本。
煤制乙二醇的全套工藝流程包括:由煤經過水煤氣變換得到CO原料氣,CO催化偶聯合成草酸酯,草酸酯加氫制得乙二醇。其中由水煤氣變換得到的CO原料氣中含有一定量的H2,而H2的存在會使CO催化偶聯合成草酸酯的催化劑中毒失活。因此,為保證CO催化偶聯合成草酸酯工藝的進行,將CO原料氣中H2雜質脫除至100ppm以下具有重要意義。目前最有效的的手段是通過選擇性氧化的脫氫工藝脫除CO原料氣中的H2。CO原料氣的選擇氧化脫氫反應通常使用專利CN104162443A公開的一種催化劑,該催化劑以貴金屬Pd作為活性組分,以氯作為助劑。該催化劑在選擇氧化脫除CO氣體中H2的過程中經過長時間的使用后,由于催化劑表面的貴金屬Pd顆粒的嚴重團聚和氯物種流失等原因,導致催化劑活性以及選擇性下降。
目前,針對于貴金屬催化劑再生方法的報道已有不少。例如,專利CN201410430746.6提供的一種煤制乙二醇過程失活鈀催化劑在線再生方法,采用氧化還原的方法對催化劑進行再生活化處理。該方法只應用由于貴金屬Pd顆粒團聚導致的催化性能下降的催化劑,而對于氯物種的流失導致催化劑性能下降的催化劑沒有再生效果。專利CN103801330提供了一種低碳烷烴脫氫催化劑的再生方法,將再生催化劑高溫炭燒后,在490-550℃用鹽酸或者含氯的有機物氯化處理,然后再進行高溫氧化還原處理。專利CN201610027798提供了一種針對CO原料氣脫氫工藝使用的氯作助劑的貴金屬Pd催化劑的活化再生方法,采用將催化劑在鹽酸溶液中先浸漬一定時間,再在氧化劑作用下氧化的活化再生方法。以上專利的再生方法需要引入含氯的有機物或者酸處理催化劑,而脫除CO氣體中H2反應為氣固反應,裝置為固定床反應裝置,若用上述有機物或酸對失活催化劑再生,需要將催化劑從反應管中拆卸出來,或者對反應裝置進行改造,增加液體進樣泵以及汽化室等額外裝置,進而增加再生成本。
因此,針對上述問題,需要開發一種應用于CO氣體脫氫應后失活Pd催化劑的原位再生方法,從而實現失活催化劑在反應裝置上的原位再生,簡化催化劑再生的步驟,這對工業生產具有重要意義。
技術實現要素:
本發明的目的在于提供一種CO脫氫凈化用Pd催化劑失活后的原位再生方法,該方法簡單易操作,能實現在CO氣體脫氫的反應裝置上的原位再生。
本發明所述的催化劑再生方法,具體步驟如下:
A.在CO氣體脫氫的反應裝置上,將反應管中失活催化劑在100-500℃惰性氣體中吹掃3-12h,脫除催化劑表面的吸附物;惰性氣體的空速為500-1000h-1。
B.將步驟A吹掃后的催化劑在200-700℃含氧氣氛中氧化處理1-12h,含氧氣氛的空速為100-1000h-1;以脫除催化劑上的積炭,氧化催化劑表面已團聚的Pd顆粒。
所述的含氧氣氛為純O2氣體或O2與惰性氣體的混合氣,其中O2的含量為50-100%。
C.將步驟B氧化后的催化劑在400-700℃含氯氣氛中氯化處理1-5h,含氯氣氛的空速為100-1000h-1;使催化劑表面氧化的Pd顆粒氯化、分散。
所述的含氯氣氛為HCl氣體或者Cl2與惰性氣體的混合氣,Cl的含量為5-10%。
D.將步驟C氯化后的催化劑在400-600℃還原氣氛中脫氯處理1-5h,還原氣氛的空速為100-1000h-1;以脫除催化劑表面的多余的Cl;
所述的還原氣氛為純H2氣體或H2與惰性氣體的混合氣,H2的含量為50-100%。
E.將步驟D脫氯處理后的催化劑在惰性氣體氛圍中降至室溫;惰性氣體的空速為500-1000h-1。
上述所有步驟中所述的惰性氣氛為N2、He或Ar中的一種。
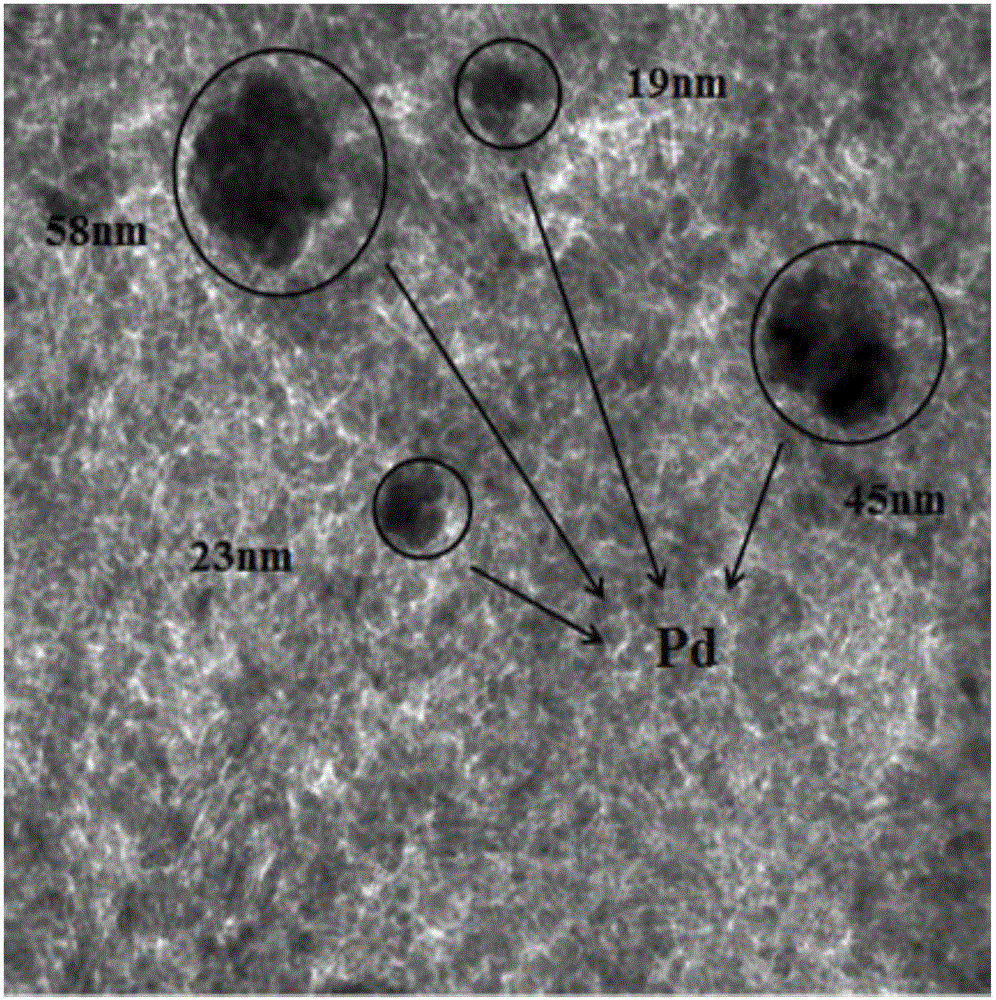
對再生前、后狀態的催化劑進行透射電鏡表征,結果見圖1、2。由圖1可見再生前的催化劑中Pd的團聚非常明顯,Pd顆粒較大;圖2可見再生后的催化劑中團聚的Pd明顯再分散,Pd的顆粒較小。
采用靜態CO化學吸附法對再生前、后催化劑進行性能測試,結果顯示Pd的分散度從8.1%提高到25.7-40.5%,略低于新鮮催化劑的分散度46.3%;Pd的顆粒尺寸從15.1nm下降到2.5-4.1nm,略高于新鮮催化劑的顆粒尺寸2.2nm。
分別將再生前、后的催化劑于固定床反應器中進行CO氣體脫氫凈化實驗,評價條件為:原料氣H2含量為1.5%,反應空速為2000h-1,O2和H2的體積比為1.5,壓力為0.2MPa,反應溫度為150℃。評價結果顯示,再生后的催化劑反應殘余的H2從4576ppm減小至100ppm以下,選擇性從58.5%提高到63.7-69.8%,略低于新鮮催化劑的80.2%。
本發明的有益效果是:再生過程在反應裝置上原位發生,無需對反應裝置進行改動,也不需要額外的再生裝置;再生效果好,處理后催化劑的Pd活性組分可以較好地分散于載體上,再生后的催化劑的選擇性可以恢復至新鮮催化劑選擇性的80%以上,H2可以脫除至100ppm以下,實現失活催化劑的原位再生。
附圖說明
圖1為實施例1中催化劑再生前的透射電鏡照片。
圖2為實施例1中催化劑再生后的透射電鏡照片。
具體實施方式
實施例1:
在CO氣體脫氫反應裝置中,將N2通入裝有失活催化劑的反應管中,在100℃下吹掃12h,N2的空速為1000h-1;將反應溫度升至700℃,通入O2與N2的混合氣氧化處理12h,混合氣的空速為1000h-1,O2的含量為50%;氧化處理完后在此溫度下,通入HCl與N2的混合氣氯化處理5h,混合氣的空速為1000h-1,HCl氣體的含量為5%,;氯化處理完后,將反應裝置降溫至400℃,通入H2與N2的混合氣還原5h,混合氣的空速為1000h-1,H2的含量為50%;還原完后通入N2將反應裝置降至室溫,N2空速為1000h-1,完成再生。
將再生后的催化劑進行性能評價,反應后殘余H2含量為54ppm,選擇性為65.3%。通過靜態CO化學吸附檢測,再生后催化劑的Pd的分散度為38.2%,Pd的平均顆粒尺寸為2.9nm。
實施例2:
同實例1的再生過程,不同之處在于處理過程中的惰性氣體He代替N2處理3h,He的空速為500h-1;氧化條件為:T=200℃下,空速為100h-1的純O2氧化處理1h代替T=700℃下,空速為1000h-1的O2含量為50%的混合氣處理12h。
將再生后的催化劑進行性能評價,反應后殘余H2含量為32ppm,選擇性為69.8%。通過靜態CO化學吸附檢測,再生后催化劑的Pd的分散度為40.5%,Pd的平均顆粒尺寸為2.5nm。
實施例3:
同實施例1的再生過程,不同之處在于處理過程中的惰性氣體Ar代替N2;氯化條件為:空速為100h-1的HCl含量為10%的混合氣氯化處理1h代替空速為1000h-1的HCl含量為5%的混合氣氯化處理5h。
將再生后的催化劑進行性能評價,反應后殘余H2含量為76ppm,選擇性為63.7%。通過靜態CO化學吸附檢測,再生后催化劑的Pd的分散度為25.7%,Pd平均顆粒尺寸為4.1nm。
實施例4:
同實施例1的再生過程,不同之處在于氯化條件為:T=400℃下Cl2含量為5%的混合氣氯化處理1h代替T=700℃下HCl氣體含量為5%的混合氣氯化處理5h。
將再生后的催化劑進行性能評價,反應后殘余H2含量為98ppm,選擇性為64.6%。通過靜態CO化學吸附檢測,再生后催化劑的Pd的分散度為29.9%,Pd的平均顆粒尺寸為3.5nm。
實施例5:
同實施例1的再生過程,不同之處在于還原條件為:T=600℃下空速為100h-1的純H2還原處理1h代替T=400℃下空速為1000h-1的含量為50%的H2混合氣還原處理5h。
將再生后的催化劑進行性能評價,反應后殘余H2含量為63ppm,選擇性為67.1%。通過靜態CO化學吸附檢測,再生后催化劑的Pd的分散度為35.5%,Pd平均顆粒尺寸為3.3nm。
比較例1:
將新鮮的催化劑采用實施例1的評價條件進行性能評,反應后殘余H2含量為12ppm,選擇性為80.2%。通過靜態CO化學吸附檢測,新鮮催化劑的Pd的分散度為46.3%,Pd平均顆粒尺寸為2.2nm。
比較例2:
將實施例1所用的失活后未再生的催化劑采用實施例1的評價條件進行性能評價,反應后殘余H2含量為4576ppm,選擇性為58.5%。通過靜態CO化學吸附檢測,失活后催化劑的Pd的分散度為8.1%,Pd平均顆粒尺寸為15.1nm。
- 還沒有人留言評論。精彩留言會獲得點贊!