氧化亞鈷納米晶?石墨烯復合材料、其制備方法及應用與流程
本發明涉及材料制備領域,特別涉及一種氧化亞鈷納米晶-石墨烯復合材料、其制備方法及應用。
背景技術:
氧化亞鈷(CoO)作為典型的過渡金屬氧化物,與其它過渡金屬氧化物,如MnO、NiO、FeO等一樣,具有特殊結構和性質。因此,從40年代以來,就引起了人們的廣泛關注和研究。CoO有兩種典型的穩定存在的物相,即呈深黃色的rock-salt相(空間群為Fm3m)和呈綠色的wurtzit相(空間群為P63mc)。CoO是優良的磁性材料,在它的尼爾溫度(298K)附近發生順磁和反磁體的轉變。塊狀的CoO材料是絕緣的反鐵磁性材料,但是納米結構的CoO(納米CoO)材料展現出良好的鐵磁性或超順磁性。納米CoO材料具有更好的物理和化學性能,其在鋰離子電池、電磁波吸收、催化、氣體傳輸及磁數據存儲設備等方面均具有廣泛的應用。
在納米CoO材料的諸多應用中,電磁波吸收性能、磁性能及催化性能是研究的熱點。一方面,在吸波材料的研究和應用上,磁性能良好的納米材料發揮主導作用。另一方面,CoO廣泛應用于催化領域,良好的磁性能可以拓寬其在催化領域的應用,有利于催化劑的分離和再利用。
然而,在現有的納米CoO材料中,由于CoO納米晶是磁性納米粒子,很容易團聚,嚴重影響了納米CoO材料的磁性能,進而對納米CoO材料的電磁波吸收性能及催化性能造成了不良影響。
技術實現要素:
本發明實施例公開了一種氧化亞鈷納米晶-石墨烯復合材料、其制備方法及應用,以解決現有的納米CoO材料中CoO納米晶容易團聚的問題。技術方案如下:
第一方面,本發明實施例提供了一種氧化亞鈷納米晶-石墨烯復合材料,所述石墨烯呈片狀,且所述氧化亞鈷納米晶分散在所述石墨烯上,所述氧化亞鈷納米晶的粒徑為5-8nm,平均粒徑為6nm。
其中,所述氧化亞鈷納米晶為面心立方結構。
第二方面,本發明實施例提供了該復合材料的制備方法,包括:
將氧化石墨加入到油胺中并進行分散處理,得到混合液;
將乙酰丙酮鈷及十八胺加入至所述混合液中,在攪拌狀態下將所述混合液加熱至120-140℃,保溫30分鐘以上,然后繼續在攪拌狀態下,加熱至290-310℃,保溫2小時以上;
加入有機溶劑將反應猝停,分離出反應產物,并對所述反應產物進行洗滌及干燥處理;
其中,所述乙酰丙酮鈷與所述氧化石墨比例為1mol:(20-40)g,所述氧化石墨與所述十八胺的質量比為1:(12.5-50),所述油胺與所述氧化石墨的比例為1L:1g。
優選的,所述分散處理為超聲分散處理。
優選的,所述加熱至290-310℃具體為:加熱至290℃。
優選的,所述有機溶劑為乙醇。
優選的,所述洗滌處理為:用正己烷及丙酮進行交替洗滌處理。
第三方面,本發明實施例還提供了該復合材料在電磁波吸收領域及催化領域的應用。
可見,本方案中以石墨烯為基底,通過熱分解法一步還原得到氧化亞鈷納米晶-石墨烯復合材料,CoO納米晶分散均勻,無團聚現象,從而使該復合材料具有良好的磁性能,同時改善了該復合材料的電磁波吸收性能及催化性能。
附圖說明
為了更清楚地說明本發明實施例或現有技術中的技術方案,下面將對實施例或現有技術描述中所需要使用的附圖作簡單地介紹,顯而易見地,下面描述中的附圖僅僅是本發明的一些實施例,對于本領域普通技術人員來講,在不付出創造性勞動的前提下,還可以根據這些附圖獲得其他的附圖。
圖1為氧化石墨(GO)及本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料的XRD圖;
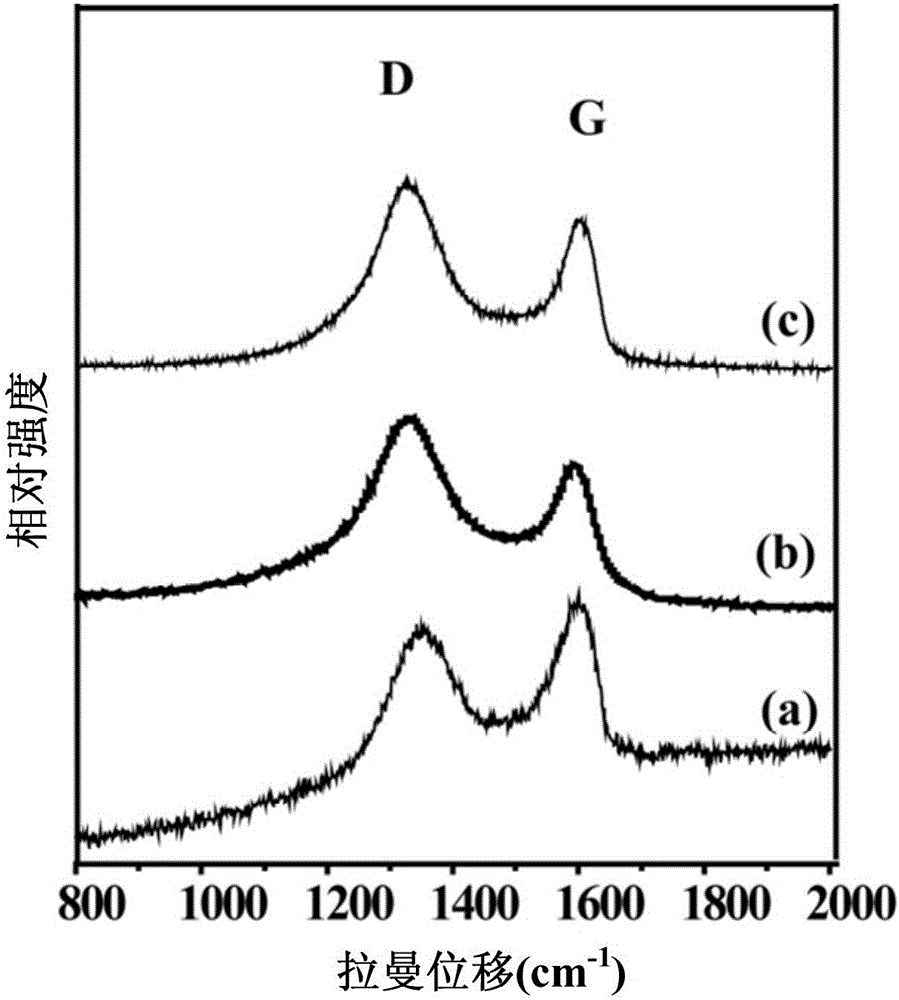
圖2為氧化石墨(GO)、石墨烯(GN)及本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料的拉曼光譜圖;
圖3為氧化石墨(GO)及本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料的XPS譜圖;
圖4為本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料的電子顯微鏡圖;
圖5為本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料的微波反射率損耗值與樣品厚度、頻率的關系圖;
圖6為本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料的磁滯回線圖;
圖7為本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料催化還原CO2的電流密度與電位的關系圖。
具體實施方式
本發明實施例公開了一種氧化亞鈷納米晶-石墨烯復合材料,其中,石墨烯(GN)呈二維片層狀,氧化亞鈷(CoO)納米晶分散在石墨烯的片層上。該氧化亞鈷納米晶-石墨烯復合材料中的CoO納米晶為面心立方結構,粒徑為5-8nm,平均粒徑約為6nm。
本發明實施例還公開了該氧化亞鈷納米晶-石墨烯復合材料的制備方法,可以包括以下步驟:
將氧化石墨加入到油胺中并進行分散處理,得到混合液;
將乙酰丙酮鈷及十八胺加入至所述混合液中,在攪拌狀態下將所述混合液加熱至120-140℃,保溫30分鐘以上,然后繼續在攪拌狀態下,加熱至290-310℃,保溫2小時以上;
加入有機溶劑將反應猝停,分離出反應產物,并對所述反應產物進行洗滌及干燥處理;
其中,所述乙酰丙酮鈷與所述氧化石墨比例為1mol:(20-40)g,所述氧化石墨與所述十八胺的質量比為1:(12.5-50),所述油胺與所述氧化石墨的比例為1L:1g。
石墨烯是由碳原子構成的只有一層原子厚度的二維晶體,它的晶格是由六個碳原子圍成的六邊形。石墨烯是已知的,目前最薄、最堅硬的納米材料,其表面積大,質量輕,導電性極好,具有良好的柔韌性和耐腐蝕性,使其可以做為基底材料與其他材料進行復合,本發明中將石墨烯作為基底材料與CoO納米晶進行復合,CoO納米晶能夠高度分散地生長在石墨烯的二維片層上。
在實際制備該氧化亞鈷納米晶-石墨烯復合材料時,可以先將氧化石墨加入到油胺中并進行分散處理,得到混合液。具體的,油胺與氧化石墨的比例可以為1L:1g。該分散處理可以采用超聲分散、機械攪拌分散處理等分散方式,優選為超聲分散處理。當該混合液中不存在懸浮物,得到顏色均勻的棕色混合溶液時,說明該混合液已經分散均勻,便可以停止分散處理。需要說明的是,該分散處理為本領域常用的處理方式,在此不做具體說明。
進一步需要說明的是,氧化石墨可以從市場購得,也可以通過現有的制備方法制得,本領域技術人員可以根據實際需要進行選擇,在此不做具體限定。本發明中所使用的氧化石墨通過下述改進的Hummers方法制得,采用該方法制備的氧化石墨氧化程度高,分散效果好,更利于氧化亞鈷納米晶-石墨烯復合材料的制備。
具體制備方法如下:
稱取2.5g石墨粉,2.5g硝酸鈉和115mL濃硫酸,置于冰水浴中,在攪拌狀態下緩慢加入15g KMnO4;大約15分鐘后,撤去冰水浴,放入35℃水浴中,緩慢加入230mL蒸餾水,產物由黑色變為褐色;然后放入98℃油浴中,保溫15分鐘后,撤去油浴,加入700mL溫水,攪拌狀態下加入50mL雙氧水,產物變為金黃色;對產物進行過濾處理后,用質量百分數為5%的稀HCl溶液洗滌,再用蒸餾水洗滌5-10次,直到濾液中無SO42-為止;將所得產物于70℃干燥箱中進行干燥處理,得到氧化石墨。
在得到上述分散均勻的混合液后,將乙酰丙酮鈷及十八胺加入至該混合液中,在攪拌狀態下將該混合液加熱至120-140℃,保溫30分鐘以上,然后繼續在攪拌狀態下,加熱至290-310℃,保溫2小時以上。具體的,乙酰丙酮鈷與氧化石墨比例可以為1mol:(20-40)g,優選為1mol:20g。氧化石墨與十八胺的質量比可以為1:(12.5-50),優選為1:12.5。
其中,十八胺作為限域劑和還原劑,一方面用于防止CoO納米晶生長不均勻,有利于形成單分散的CoO納米晶,另一方面,十八胺作為一種還原劑,可以避免Co3+的生成,同時還原氧化石墨為石墨烯,利于復合材料的生成。發明人發現氧化石墨與十八胺的質量比為1:12.5時更有利于復合材料的生成。
將該混合液加熱至120-140℃,保溫30分鐘以上,可以使CoO納米晶成核并生長,可以理解的是,保溫時間越長,CoO納米晶成核及生長的效果越好,但是保溫時間過長不僅會使制備效率降低,還會增加制備復合材料的成本。發明人發現將該混合液加熱至120℃,保溫30分鐘時,制備效率高且成本較低,因此優選為將該混合液加熱至120℃,保溫30分鐘。
進一步,在攪拌狀態下進行加熱及保溫處理可以使乙酰丙酮鈷、氧化石墨及十八胺在油胺中混合的更加均勻,進而使反應更加均勻,有利于復合材料的生成。優選的,該攪拌狀態可以為磁力攪拌狀態。當然也可以為其他攪拌方式,只要不影響復合材料的生成且能夠達到攪拌目的即可,在此不做具體限定。
同理,在上述保溫處理后,為了進一步為CoO納米晶的生長提供有利環境,可以繼續在攪拌狀態下,加熱至290-310℃,保溫2小時以上。考慮到制備效率及成本的要求,優選為加熱至290℃,保溫2小時。該攪拌狀態也可以為磁力攪拌狀態,當然也可以為其他攪拌方式,只要不影響復合材料的生成且能夠達到攪拌目的即可,在此不做具體限定。
為了制備得到尺寸均勻的CoO納米晶,可以在上述第二次保溫處理后將反應猝停。具體可以采用加入有機溶劑的方式,加入有機溶劑后可以使混合液迅速降溫,進而將反應猝停,該有機溶劑優選為乙醇。
將反應猝停后,需要進行分離處理,即將所制備的氧化亞鈷納米晶-石墨烯復合材料與混合液分離開,可以采用離心或過濾等方式進行分離處理,優選為離心分離。分離出產物后,可以對產物用正己烷及丙酮進行交替洗滌處理,然后進行干燥處理,得到氧化亞鈷納米晶-石墨烯復合材料。對于洗滌次數,可以由本領域技術人員根據實際產物情況進行選擇,例如可以為3次或4次,在此不做具體限定。對于干燥處理可以將產物于40℃進行真空干燥處理,當然也可以采用其它干燥處理方式,在此不做具體限定。
本發明還提供了由上述方法制得的氧化亞鈷納米晶-石墨烯復合材料用于吸波領域以及催化領域的用途。
需要說明的是,上述分散、攪拌、離心、過濾、洗滌及干燥處理均為本領域常用的處理方法,本領域技術人員可以根據實際制備情況進行操作,在此不做具體限定及說明。
進一步需要說明的是,在制備該復合材料過程中所用到的原料,均可以在市場上購得或自制,在此不做具體限定。
下面將結合本發明實施例對本發明實施例中的技術方案進行描述,顯然,所描述的實施例僅僅是本發明一部分實施例,而不是全部的實施例。基于本發明中的實施例,本領域普通技術人員在沒有作出創造性勞動前提下所獲得的所有其他實施例,都屬于本發明保護的范圍。
實施例1
將40mg氧化石墨(GO)加入到40mL油胺中,進行超聲分散約2小時后,得到分散均勻的棕色混合液;
將2mmol乙酰丙酮鈷(Co(acac)2)和0.5g十八胺加入至上述棕色混合液中,在磁力攪拌狀態下加熱至120℃,保溫30分鐘,然后繼續在磁力攪拌狀態下加熱至290℃,保溫2小時;
加入20mL乙醇將反應猝停,然后進行離心處理,分離出反應產物,并用正己烷、丙酮交替洗滌3次后,將產物于40℃進行真空干燥。
實施例2
將40mg氧化石墨(GO)加入到40mL油胺中,進行超聲分散約2小時后,得到分散均勻的棕色混合液;
將1mmol乙酰丙酮鈷(Co(acac)2)和1.4g十八胺加入至上述棕色混合液中,在磁力攪拌狀態下加熱至135℃,保溫40分鐘,然后繼續在磁力攪拌狀態下加熱至300℃,保溫3小時;
加入20mL乙醇將反應猝停,然后進行離心處理,分離出反應產物,并用正己烷、丙酮交替洗滌4次后,將產物于40℃進行真空干燥。
實施例3
將30mg氧化石墨(GO)加入到30mL油胺中,進行超聲分散約2小時后,得到分散均勻的棕色混合液;
將1mmol乙酰丙酮鈷(Co(acac)2)和1.5g十八胺加入至上述棕色混合液中,在磁力攪拌狀態下加熱至140℃,保溫60分鐘,然后繼續在磁力攪拌狀態下加熱至310℃,保溫4小時;
加入20mL乙醇將反應猝停,然后進行離心處理,分離出反應產物,并用正己烷、丙酮交替洗滌5次后,將產物于40℃進行真空干燥。
表征與分析
1、X射線衍射(X-ray Diffraction,XRD)分析
采用荷蘭PAN alytical公司生產的X射線粉末衍射儀(型號:X Pert PRO MPD)對本發明制備的氧化石墨(GO)及本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料進行表征測得的XRD圖如圖1所示。
圖1中(a)為本發明制備的氧化石墨的XRD圖,由圖可見,通過對石墨進行氧化而得到的GO的層間距為(原始石墨約為),同時在2θ=10.8°位置有一個典型的衍射峰,而原始石墨的典型衍射峰(2θ=26.5°)消失,說明獲得的GO已經被有效的氧化。
圖1中(b)為本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料的XRD圖,由圖可見,本發明實施例制備的氧化亞鈷納米晶-石墨烯復合材料的衍射峰位于2θ=36.5°、42.3°、61.5°、73.6°及77.0°,通過與PDF卡號為48-1719的標準卡片比對,證明獲得的是CoO。進一步指標化可知,樣品的衍射峰分別對應于(111)、(200)、(220)、(311)及(222)晶面,層間距分別為0.246、0.213、0.151、0.128及0.123nm,對應面心立方結構,空間群是Fm3m。同時,在本發明實施例制備的氧化亞鈷納米晶-石墨烯復合材料中,GO的衍射峰消失,證明不存在GO,這可能與GO被還原成了無定形的石墨烯(GN)有關。本發明實施例制備的氧化亞鈷納米晶-石墨烯復合材料的XRD衍射峰明顯寬化,推測得到的CoO納米晶尺寸較小。
2、拉曼光譜分析
采用Horiba Jobin Yvon公司生產的拉曼光譜儀(型號:LavRAMAramis)對本發明制備的氧化石墨(GO)、本發明所用的石墨烯(GN)及本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料進行表征測得的拉曼光譜圖如圖2所示。其中,圖2中(a)為本發明制備氧化石墨的拉曼光譜圖,圖2中(b)為本發明所用的石墨烯的拉曼光譜圖,圖2中(c)為本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料的拉曼光譜圖。
拉曼光譜圖被廣泛用于檢測碳材料的結構特征。從天然石墨到氧化石墨再漸變到石墨烯,材料的結構發生了巨大變化,而這種變化能夠從拉曼光譜圖中表現出來。在碳材料的拉曼光譜中,分別把拉曼位移在1345cm-1和1570cm-1附近對應的峰,稱為D帶和G帶,它們是碳原子晶體的拉曼特征峰。D帶對應于石墨無序的sp3雜化碳原子振動,與混亂度和晶格缺陷有關,G帶對應于二維的六方晶格sp2雜化的碳原子平面振動,與材料的堆垛結構相關。
從圖2中(a)可以看出,本發明制備的氧化石墨的D帶和G帶的強度比,即ID/IG約為0.96:1,已知石墨的ID/IG約為0.27:1,可見隨著含氧官能團的引入和sp2雜化碳原子區域被破壞,氧化石墨晶格缺陷增大,所以其ID/IG比石墨的ID/IG大。從圖2中(b)可以看出,石墨烯的ID/IG約為1.21:1,與本發明制備的氧化石墨相比,石墨烯的ID/IG有所增大,可能是因為還原過程中,石墨烯的片層變小所致。從圖2中(c)可以看出,本發明實施例制備的氧化亞鈷納米晶-石墨烯復合材料的ID/IG約為1.20:1,表明在本發明實施例制備的氧化亞鈷納米晶-石墨烯復合材料中氧化石墨已經被還原了。
3、X射線光電子能譜(X-ray Photoelectron Spectroscopy,XPS)分析
采用XPS光譜儀(型號:ESCALAB 250Xi)對本發明制備的氧化石墨(GO)及本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料的表面組成及鈷的價態進行表征測得的XPS譜圖如圖3所示。
圖3中(a)為本發明制備的氧化石墨的C1s的XPS圖譜。對碳材料而言,一般288.9、287.7、286.0及284.7eV分別對應O=C-O、C=O、O-C-O及C-C的結合能。由圖(a)可見,發明制備的氧化石墨中含有大量含氧官能團。
圖3中(b)為本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料的C1s的XPS圖譜。由圖3中(b)可見,在氧化亞鈷納米晶-石墨烯復合材料中,288.9eV對應的峰消失,287.7及286.0eV左右對應的峰減弱,即本發明實施例制備的氧化亞鈷納米晶-石墨烯復合材料中幾乎不含O=C-O官能團,且O=C及O-C-O的含量很少,主要以C-C結構存在。表明本發明實施例制備的氧化亞鈷納米晶-石墨烯復合材料中,含氧官能團被有效地還原了。
圖3中(c)為本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料的Co 2p的XPS譜圖。圖中780.72eV附近對應的峰屬于鈷的2p3/2峰,同時伴隨著786.10eV附近對應的衛星峰(satellite peak)。796.73eV附近對應的峰屬于鈷的2p1/2峰,同時伴隨著802.69eV附近的對應的衛星峰。從圖中可以看出,結合能差ΔE即鈷的2p3/2峰對應的結合能796.73eV,與鈷的2p1/2峰對應的結合能780.72eV的差值為16.01eV。由于當ΔE為16eV時,表明是二價的鈷(CoO);當ΔE為15eV時,表明是三價的鈷(Co2O3);當ΔE為15.2eV時,表明是二價和三價的鈷的混合價態,例如Co3O4,所以本發明實施例制備的氧化亞鈷納米晶-石墨烯復合材料中得到的是二價鈷,即CoO,不含有三價鈷。同時,在778.10eV附近沒有對應的峰,表明本發明實施例制備的氧化亞鈷納米晶-石墨烯復合材料中不含有金屬Co單質。XPS光譜數據與XRD數據及拉曼光譜數據一致,表明氧化亞鈷納米晶-石墨烯復合材料能夠通過本發明所提供的方法制得。
4、電鏡圖像分析
圖4中(a)為采用Hitachi公司的掃描電子顯微鏡(Scanning Electron Microscope,SEM,型號:S-4800,加速電壓:5kV)對本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料進行表征測得的SEM圖。由圖可見,大片的石墨烯負載著細小的CoO納米晶,CoO納米晶在石墨烯片層上分散均勻,沒有明顯團聚現象。
圖4中(b)-(d)為采用透射電子顯微鏡對本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料進行表征測得的透射電子顯微鏡(Transmission Electron Microscope,TEM)圖。從圖4中(b)-(d)可以看出,本發明實施例制備的氧化亞鈷納米晶-石墨烯復合材料中的CoO納米晶為球形顆粒,CoO納米晶高密度、單分散地分布在石墨烯的片層上。從圖4中(c)及(d)可以清楚地看到石墨烯的邊緣,幾乎沒有石墨烯片層的疊加現象,說明發明實施例制備的氧化亞鈷納米晶-石墨烯復合材料中的石墨烯很薄,且沒有團聚現象。同時,CoO納米晶牢牢地生長在石墨烯片層上。
圖4中(e)及(f)為采用高分辨電子顯微鏡(High Resolution Transmission Electron Microscopy,HRTEM,型號:JEM-2010,加速電壓:200kV)對本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料進行表征測得的HRTEM圖。由于石墨烯比較薄,容易出現褶皺和團聚,但是從圖4中(e)結合圖4中(c)及(d)可以看出,本發明制備的氧化亞鈷納米晶-石墨烯復合材料中,CoO納米晶的粒徑約為5-8nm,平均粒徑約為6nm。且沒有發現石墨烯團聚和大面積裸露的現象,也沒有散落在石墨烯納米片外的CoO納米晶,而是CoO納米晶單分散地生長在薄薄的石墨烯納米片上,表明生長在石墨烯上的CoO納米晶起到了阻止石墨烯團聚的作用。這也與XRD數據一致,XRD圖中沒有石墨烯重疊的峰。從圖4中(f)可以看出CoO納米晶清晰的晶格,大部分CoO納米晶的晶格間距為0.248nm,與CoO的(111)面對應,說明立方相的CoO納米晶沿著(111)晶面生長。
圖4中(g)為本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料的選區電子衍射(Selected Area Electron Diffraction,SAED)圖,從圖4中(g)可以看出,本發明實施例制備的氧化亞鈷納米晶-石墨烯復合材料具有多個清晰的衍射環,通過計算得知,分別對應CoO納米晶的(111)、(200)、(220)、(311)及(222)晶面,與XRD數據相一致。
5、吸波性能結果
為評估本發明實施例制備的氧化亞鈷納米晶-石墨烯復合材料的微波吸收性能,把本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料與石蠟均勻混合(本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料在混合物中的質量百分數為60%,石蠟沒有電磁波吸收性能),組裝成一個電磁波吸收裝置,該裝置的外徑和內徑分別是7.00nm和3.04nm,采用Agilent E8362B矢量網絡分析儀進行表征。測試在固定的頻率和樣品厚度下進行,反射損耗值(RL)根據微波傳輸理論進行計算,公式如下:
RL(dB)=20log|(Zin-Z0)/(Zin+Z0)|
其中,Zin為吸波體的輸入阻抗,Z0為空氣阻抗,μr為相對磁導率,εr為相對介電常數,f為微波頻率,d為樣品厚度,c為光速。
圖5為通過上述電磁波吸收裝置測得的本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料的微波反射率損耗值與樣品厚度的關系圖。在1.0-18.0GHz頻率范圍內測試了不同樣品厚度的反射損耗值隨頻率的變化關系。當吸波材料的反射損耗值為-10dB時,表示90%的電磁波被吸收,只有10%的電磁波被反射,稱為有效吸收。從圖5可見,隨著樣品厚度的增加,出現最小損耗值的吸收頻率向低頻移動,同時吸收強度增加,吸收帶增多。通過調節樣品的厚度,樣品在低頻(1-6GHz)、中頻(7-12GHz)及高頻(13-18GHz)的反射損耗值均能夠低于-10dB。
具體的,從圖5中可以看到,當樣品厚度為2mm時,最強吸收出現在14.80GHz,反射損耗值為-8.1dB;當樣品厚度為3mm時,最強吸收出現在9.32GHz,反射損耗值為-9.5dB;當樣品厚度為4mm時,最強吸收出現在6.78GHz,反射損耗值為-9.5dB;當樣品厚度大于4mm時,出現多個最強吸收峰,如樣品厚度為5mm時,最強吸收為4.91GHz,反射損耗值為-10.5dB,以及17.49GHz,反射損耗值為-24.1dB;樣品厚度為6mm時,最強吸收為4.06GHz,反射損耗值為-12.6dB,以及14.43GHz,反射損耗值為-23.8dB;樣品厚度為7mm時,最強吸收為3.38GHz,反射損耗值為-14.3dB,以及12.05GHz,反射損耗值為-28.9dB;樣品厚度為8mm時,最強吸收為2.70GHz,反射損耗值為-15.7dB,以及10.35GHz,反射損耗值為-22.0dB。當樣品厚度為9mm時,出現三個有效吸收峰(RL<-10dB),分別出現在低頻2.36GHz,反射損耗值為-16.7dB、中頻8.99GHz,反射損耗值為-17.5dB以及高頻16.13GHz,反射損耗值為-13.7dB,即樣品厚度為9mm時在全波段都能達到有效吸收。
眾所周知,當吸波材料的反射損耗值達到-10dB時,這樣的吸波材料就具有實際的應用價值。另外,隨著吸收劑厚度的增加,其吸收強度增大,并且吸收頻帶向低頻移動,通過調節樣品的厚度,反射損耗可以覆蓋1-18GHz頻帶,證明本發明實施例制備的氧化亞鈷納米晶-石墨烯復合材料可以通過調節厚度來達到全波段吸收的實際應用價值。
6、磁性能結果
采用美國Quantum Design公司生產的磁化曲線測試儀器(型號:MPMS-s-SQUID)在外加磁場的磁飽和度約為10KOe條件下對本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料進行表征測得的磁滯回線圖(M-H回路圖)如圖6所示。其中,M表示磁化強度,H表示磁場強度。
圖6中分別為本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料在室溫(圖中以RT表示)和溫度為2K下測得的磁滯回線圖。由圖6可見,本發明制備的氧化亞鈷納米晶-石墨烯復合材料在室溫和2K溫度下都表現出鐵磁性,其飽和磁化強度分別約為8.2emu/g和8.8emu/g。該飽和磁化強度均遠大于納米花狀(粒徑為50nm)、八面體納米結構(粒徑為25nm)及納米球狀(粒徑為7nm)的CoO在5K和300K下測得的飽和磁化強度,同時,大于已報道的CoO與還原氧化石墨(RGO)形成的納米復合材料(納米粒徑為20nm)及其在加熱到300℃和400℃后測得的飽和磁化強度。本發明制備的氧化亞鈷納米晶-石墨烯復合材料的矯頑力在室溫和2K下分別約為0.29Koe和0.38KOe。由此可見,本發明制備的氧化亞鈷納米晶-石墨烯復合材料具有良好的磁性能。
7、催化性能結果
采用線性掃描伏安法測試儀器(型號:Zahner IM6)在CO2和Ar氛圍中對本發明實施例1制備的氧化亞鈷納米晶-石墨烯復合材料進行表征測得的催化CO2的電流密度與電位的關系圖如圖7所示。
由圖可見,在CO2氛圍中對應的電流密度絕對值明顯大于在Ar氛圍中對應的電流密度絕對值,這是由于在電解水的同時也在電解還原CO2,所以電流密度絕對值會比僅僅在Ar氛圍中電解水的電流密度絕對值更大。在CO2氛圍中的起峰電位明顯提前于在Ar氛圍中電解水的起峰電位。而且,隨著電位繼續升高,電流密度絕對值繼續增大,表明本發明實施例制備的氧化亞鈷納米晶-石墨烯復合材料作為催化劑,對CO2電催化還原有著非常好的催化性能。
可見,本方案中以石墨烯為基底,通過熱分解法一步還原得到氧化亞鈷納米晶-石墨烯復合材料,CoO納米晶分散均勻,無團聚現象,從而使該復合材料在具有良好的磁性能的同時,改善了該復合材料的吸波性能及催化性能。
以上對本發明所提供的氧化亞鈷納米晶-石墨烯復合材料、其制備方法及應用進行了詳細介紹。本文中應用了具體實施例對本發明的原理及實施方式進行了闡述,以上實施例的說明只是用于幫助理解本發明的方法及其中心思想。應當指出,對于本領域的普通技術人員來說,在不脫離本發明原理的前提下,還可以對本發明進行若干改進和修飾,這些改進和修飾也落入本發明權利要求的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!