一種活性炭基固體酸型非均相芬頓催化劑及其制備方法與流程
本發明屬于處理水中難降解有機污染物的應用,具體涉及一種活性炭基固體酸型非均相芬頓催化劑及其制備方法。
背景技術:
Fenton氧化技術因具有反應條件溫和,處理效率高,價格便宜,在常溫常壓下即可完成催化降解反應等優點,常用于污水處理。Fenton試劑法的實質是在酸性條件下,H2O2在Fe2+的催化作用下,產生了具有高反應活性的羥基自由基,·OH是一種氧化能力很強的自由基,具有較高的氧化還原電位,能迅速氧化廢水中的污染物,可使廢水中難于生物降解的大分子有機物裂解為易于微生物降解的小分子有機物,或者降解一般化學氧化法難以有效降解的印染廢水。但目前Fenton試劑處理廢水時需在酸性條件下進行,對反應環境的pH值要求較高(pH值在3.0左右),水處理之前需先調低pH值,處理后液體酸又難以回收;反應后催化劑不易于原料及產物分離,Fe2+的加入還會產生二次污染。
技術實現要素:
本發明的目的在于針對現有技術的不足,提供一種活性炭基固體酸型非均相芬頓催化劑及其制備方法。本發明通過將活性炭基固體酸與亞鐵離子共價鍵合制得芬頓催化劑;該催化劑解決了現有的芬頓催化劑易溶出金屬離子和液體酸根離子的問題,性能穩定,效果突出。
為實現上述發明目的,本發明采用如下技術方案:
一種活性炭基固體酸型非均相芬頓催化劑的制備方法:將活性炭基固體酸浸漬于含亞鐵離子的溶液中,持續振蕩14-20h,過濾,然后用去離子水洗滌至檢測不出亞鐵離子;將處理后的活性炭基固體酸置于真空下烘干,得到活性炭基固體酸型非均相芬頓催化劑。
所述的活性炭基固體酸是富含羧基官能團的活性炭基固體酸。
所述的活性炭基固體酸的制備方法為:將含有氨基的苯基羧酸進行重氮化處理,得到苯基羧酸重氮鹽;然后該苯基羧酸重氮鹽與活性炭在55-60℃反應,使苯基羧酸共價鍵合在活性炭上,反應結束后,將活性炭經過濾、洗滌和烘干后即得富含羧基的活性炭基固體酸。
所述的含亞鐵離子的溶液的濃度為0.5-1.0 mol/L。
活性炭基固體酸與含亞鐵離子的溶液的浸漬比為8~40g:1L。
所述的烘干的溫度為80℃,時間為12h。
所述的含亞鐵離子的溶液包括FeSO4溶液、FeCl2溶液和Fe(NO3)2溶液中的一種。
一種如上所述的制備方法制得的活性炭基固體酸型非均相芬頓催化劑。
所述的活性炭基固體酸型非均相芬頓催化劑的應用:在紫外光照射下,H2O2存在下,降解廢水中的有機污染物。
更具體的,本發明的活性炭基固體酸可按照下述文獻中的方法制備得到:(發明專利:ZL 2014101233423.2J)。
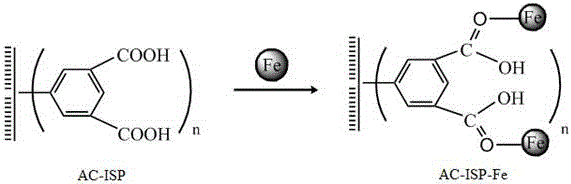
活性炭基固體酸浸漬于亞鐵離子溶液中,共價鍵合了二價鐵離子后,這點從其紅外光譜圖中可得到印證。其紅外圖譜羰基吸收峰變弱,這表明活性炭基固體酸的羧基官能團與亞鐵離子發生了共價鍵合。
本發明的有益效果在于:
1)活性炭表面增加的酸中心羧基與亞鐵離子之間是以共價鍵結合固定,溶出離子大大減少;固體催化劑本身就負載著酸,替代了液體酸,不需要再向反應設備中加入強酸,降低了設備的要求;使用過的活性炭基固體酸催化劑能重復使用;
2)本發明的制備方法簡單,僅需將活性炭基固體酸浸漬于亞鐵離子溶液中,即可得到性能穩定的催化劑,生產成本低。
附圖說明
圖1為實施例1活性炭(AC),活性炭基固體酸(AC-ISP)和活性炭基固體酸共價鍵合亞鐵離子樣品(AC-ISP-Fe,芬頓催化劑)的紅外光譜的變化情況;
圖2為實施例1活性炭基固體酸共價鍵合亞鐵離子得到的芬頓催化劑(AC-ISP-Fe)的示意圖;
圖3為實施例1活性炭,活性炭基固體酸和活性炭基固體酸共價鍵合亞鐵離子樣品XPS的變化情況。
具體實施方式
下面通過具體實施例對本發明做進一步說明,但本發明并不局限于此。
下述實施例中所述實驗方法,如無特殊說明,均為常規方法; 所述試劑和材料,如無特殊說明,均可從商業途徑獲得。
實施例1
活性炭基固體酸型芬頓催化劑制備方法如下:
在燒瓶中加入5.5 g 5-氨基間苯甲酸和3 mol/L HCl,混合加熱攪拌;上述溶液冷卻至室溫,加入12wt% NaNO2,用淀粉-KI試紙控制反應終點,在0℃ 下攪拌反應30 min,即得間苯二甲酸重氮鹽。然后加入活性炭在55℃反應90 min,過濾,依次用蒸餾水回流洗滌12 h,乙醇回流洗滌12 h,置于干燥箱110℃干燥24 h,得固體酸;
稱取4.5 g固體酸浸漬于0.25 L的0.7 mol/L的FeSO4溶液中,持續振蕩14 h,將樣品用去離子水多次洗滌,直至檢測不出亞鐵離子。將樣品置于真空下烘干,得到活性炭基固體酸芬頓催化劑。
實施例2
活性炭基固體酸型芬頓催化劑制備方法如下:
在燒瓶中加入5.5 g 5-氨基間苯甲酸和3 mol/L HCl,混合加熱攪拌;上述溶液冷卻至室溫,加入12wt% NaNO2,用淀粉-KI試紙控制反應終點,在5℃下攪拌反應30 min,即得間苯二甲酸重氮鹽。然后加入活性炭在60℃反應90 min,過濾,依次用蒸餾水回流洗滌12 h,乙醇回流洗滌12 h,置于干燥箱110℃干燥24 h,得固體酸;
稱取4.5 g固體酸浸漬于0.25 L的0.8 mol/L的FeSO4溶液中,持續振蕩14h,將樣品用去離子水多次洗滌,直至檢測不出亞鐵離子。將樣品置于真空下烘干,得到活性炭基固體酸芬頓催化劑。
實施例3
活性炭基固體酸型芬頓催化劑制備方法如下:
在燒瓶中加入5.5 g 5-氨基間苯甲酸和3 mol/L HCl,混合加熱攪拌;上述溶液冷卻至室溫,加入12wt% NaNO2,用淀粉-KI試紙控制反應終點,在2℃下攪拌反應30 min,即得間苯二甲酸重氮鹽。然后加入活性炭在58℃反應90 min,過濾,依次用蒸餾水回流洗滌12 h,乙醇回流洗滌12 h,置于干燥箱110℃干燥24 h,得固體酸;
稱取4.5 g固體酸浸漬于0.25 L的1.0 mol/L的FeSO4溶液中,持續振蕩14 h,將樣品用去離子水多次洗滌,直至檢測不出亞鐵離子。將樣品置于真空下烘干,得到活性炭基固體酸芬頓催化劑。
應用實施例1
對比普通活性炭和實施例1制得的活性炭基固體酸芬頓催化劑,考察制備的催化劑是否具有紫外光催化性能。
稱取0.1 g催化劑,直至在50 mg/L的甲基橙100 ml模式廢水中吸附直至飽和,然后加入0.4 ml 30%的H2O2,在356 nm的紫外光下攪拌反應,在460 nm位置測吸光度并計算其降解率。對活性炭和活性炭基固體酸芬頓催化劑光催化反應過程進行對比。結果表明90分鐘內活性炭去除率為2.2%,活性炭基固體酸芬頓催化劑去除率為85.3%。說明活性炭作為催化劑,在紫外光下的催化作用不大。本發明制備的催化劑與普通活性炭相比,在在紫外光下具有較好的催化性能。證明該催化劑具有紫外光催化性能。
應用實施例2
稱取0.2 g實施例1制得的催化劑,在50 mg/L的甲基橙100 ml模式廢水中吸附直至飽和,然后加入0.4ml 30%的H2O2,在356 nm的紫外光下攪拌反應,在460 nm位置測吸光度并計算其降解率。對活性炭和活性炭基固體酸芬頓催化劑光催化兩種反應過程進行對比。結果表明90分鐘內活性炭去除率為7.1%,活性炭基固體酸芬頓催化劑去除率為98.0%。說明活性炭作為催化劑,在紫外光下的催化作用不大。本發明制備的催化劑與普通活性炭相比,在在紫外光下具有較好的催化性能。證明該催化劑具有紫外光催化性能。
應用實施例3
稱取0.1 g催化劑,在50 mg/L的甲基橙100 ml模式廢水中吸附直至飽和,然后加入0.4 ml 30%的H2O2,在356 nm的紫外光下攪拌反應,在460 nm位置測吸光度并計算其降解率。對活性炭和活性炭基固體酸芬頓催化劑光催化反應過程進行對比。結果表明90分鐘內活性炭去除率為5.1%,活性炭基固體酸芬頓催化劑去除率為95.5%。說明活性炭作為催化劑,在紫外光下的催化作用不大。本發明制備的催化劑與普通活性炭相比,在在紫外光下具有較好的催化性能。證明該催化劑具有紫外光催化性能。
以上所述僅為本發明的較佳實施例,凡依本發明申請專利范圍所做的均等變化與修飾,皆應屬本發明的涵蓋范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!