使用非均相催化劑的一鍋法方法與流程
相關申請的交叉引用
本申請要求2014年11月19日提交的歐洲申請號14193925.6的優先權,出于所有目的將所述申請的全部內容通過援引方式并入本申請。
本發明涉及一種用于使有機化合物一鍋法氫化并且脫水或異構化的方法。
背景技術:
目前使用逐步方法生產若干重要的市場產品、精細或商品化學品,這些方法要求單獨的反應單元、純化步驟和經常劇烈的反應條件,具有嚴重的經濟和環境缺點。對于在工業規模上容易可應用的并且能夠使一鍋法級聯/串聯反應順序提供將基礎底物更快、更環保且成本效益的全部轉化成更復雜化學品的方法仍然感覺到一種需求。
乙酰丙酸是己糖酸水解的眾所周知的產物,并且是從纖維素原料廉價地獲得的。因此,它是在生產有用的5-碳化合物如甲基四氫呋喃及衍生物和5-甲基-二氫-呋喃-2-酮(還被稱為γ-戊內酯)中有吸引力的起始材料。
us2,786,852a(桂格燕麥公司(quakeroatscompany))3/26/1957披露了使用還原的氧化銅催化劑在從175℃至250℃的溫度下從懸浮在氫氣氣體流中的以汽化形式的乙酰丙酸生產5-甲基-二氫-呋喃-2-酮。
us5,883,266a(巴特爾紀念研究所(battellememorialinstitute))3/16/1999披露了使用雙金屬催化劑在200℃或250℃的反應溫度下在100atm的操作壓力下從乙酰丙酸制備多種產物(包括5-甲基-二氫-呋喃-2-酮)。
us4,420,622a(斯塔密卡邦公司(stamicarbon))12/13/1983涉及使用c8烴乙酰丙酸酯制備5-烷基-丁內酯,其中具有最高達4個碳原子的側鏈烷基取代基作為起始原料,其中該反應是在從150℃至325℃的溫度下在氣相中并且在由周期表的第viii族或第ib族的金屬構成的氫化催化劑的存在下用氫氣進行的。
us20040254384a(杜邦公司(dupont))12/16/2004披露了在超流體溶劑存在下使用金屬催化劑將乙酰丙酸轉化為γ-戊內酯。
本發明的目的是提供一種用于在相對溫和的條件下將可容易獲得的起始材料(如乙酰丙酸)轉化成高級中間體如γ-戊內酯及其衍生物的方法。本發明的另一個目的是提供一種用于此類轉化的方法,該方法可以容易地被擴展到工業生產并且是成本效益的且環保的。
發明概述
這些目的是通過本發明借助于一種用于使含有至少一個c=c、c=o、縮醛、1,2-二醇或半縮醛部分的底物(i)一鍋法氫化并且酸催化異構化或脫水以獲得產物(ii)的方法解決的,其中所述方法包括在催化劑(iii)的存在下使該底物與氫氣(h2)反應的步驟,所述催化劑包含負載在包含至少一種氟化聚合物(p)的材料上的粒徑低于50nm的過渡金屬顆粒,其中聚合物(p)帶有-so2x官能團,x選自x'和om,x'選自由f、cl、br和i組成的組;并且m選自由h、堿金屬和nh4組成的組。
諸位發明人發現本發明的一鍋法方法以高產率提供了高級中間體,其中很少或沒有副產物,并且該固體催化劑可以容易地在若干個循環中被回收和再使用而沒有催化活性的損失。
與現有技術的方法不同,用本發明的一鍋法方法獲得了高催化劑生產率,而沒有加入強酸,導致更清潔且更環保的方法和更長的催化劑生命周期。
在現有技術的方法中,需要額外的下游加工步驟來去除從催化劑中瀝濾的金屬和/或純化反應產物,苛刻的反應條件(高的溫度和氫氣壓力、有毒試劑)。值得注意地,發現在本發明的方法中,負載在該氟化聚合物上的金屬原子不會釋放到反應介質中,因此純化步驟不是必要的。
附圖簡要說明
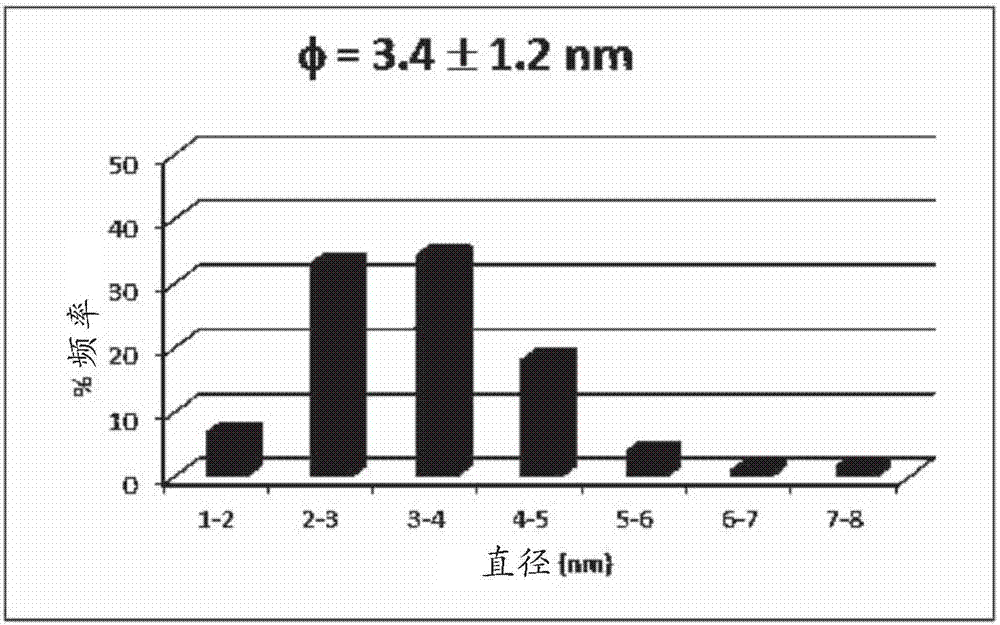
圖1示出了根據本發明的組合物的tem圖像(金屬=ru、平均d50粒徑=3.4微米,載體=
圖2示出了如圖1中示出的組合物的粒徑分布。
實施方式的說明
在一個實施例中,本發明提供了一種用于在雙功能催化劑存在下使底物氫化并且脫水/異構化的一鍋法方法。
如在此使用的,“一鍋法”指示一種方法,其中在單個反應器單元中從底物開始進行兩種或更多種化學轉化,而沒有任何反應中間體的離析或分離。
所述催化劑包含在包含至少一種帶有-so2x官能團的氟化聚合物(p)的載體上的活性金屬顆粒,其中x是如以上定義的。
已經發現包含磺酸離子交換基團的氟化聚合物(也稱為離聚物或pfsa),主要由于其離子傳導特性,被廣泛用在制造用于電化學裝置諸如電解池和燃料電池的電解質膜中。值得注意的實例是例如質子交換膜(pem)燃料電池,其使用氫氣作為燃料并且使用氧氣或空氣作為氧化劑。
如在此使用的術語“氟化聚合物”或“氟聚合物”指的是或者完全地或者部分地氟化的(即,其中烴結構的氫原子的所有或僅一部分已經被氟原子替換)化合物(例如聚合物、單體等)。優選地,術語“氟化的”指的是包含比氫原子更高比例的氟原子的化合物,更優選地指的是完全不含氫原子,即其中所有的氫原子已經被氟原子替換的化合物(全氟化合物)。
合適的包含-so2x’官能團的氟化聚合物是下述那些聚合物,它們包括衍生自至少一種包含至少一個-so2x’官能團的烯鍵式不飽和氟化單體(如下文定義的單體(a))的重復單元和衍生自至少一種烯鍵式不飽和氟化單體(如下文定義的單體(b))的重復單元。
關于(a)和(b)這兩種類型的單體,短語“至少一種單體”在此用來指示每一種類型的一種或多于一種單體可以存在于該聚合物中。下文中術語單體將用來指的是給定類型的一種和多于一種單體兩者。
合適的單體(a)的非限制性實例是:
具有下式的磺酰鹵氟烯烴:cf2=cf(cf2)pso2x’,其中p是在0與10之間的整數,優選在1與6之間,更優選p是等于2或3,并且其中優選地x’=f;
具有下式的磺酰鹵氟乙烯基醚:cf2=cf-o-(cf2)mso2x’,其中m是在1與10之間、優選在1與6之間、更優選在2與4之間的整數,甚至更優選m等于2,并且其中優選x’=f;
具有下式的磺酰鹵氟烷氧基乙烯基醚:cf2=cf-(ocf2cf(rf1))w-o-cf2(cf(rf2))yso2x’
其中w是在0與2之間的整數,rf1和rf2,彼此相同或不同,獨立地是f、cl或c1-c10氟烷基,任選地被一個或多個醚氧原子取代,y是在0與6之間的整數,優選地w是1,rf1是-cf3,y是1并且rf2是f,并且其中優選x’=f;
具有式cf2=cf-ar-so2x’的磺酰鹵芳香族氟烯烴,
其中ar是c5-c15芳香族的或雜芳香族的取代基,并且其中優選x’=f。
優選地,單體(a)選自磺酰基氟化物的組,即其中x’=f。更優選地,單體(a)選自具有式cf2=cf-o-(cf2)m-so2f的氟乙烯基醚的組,其中m是在1與6之間、優選地在2與4之間的整數。甚至更優選地,單體(a)是cf2=cfocf2cf2-so2f(全氟-5-磺酰基氟化物-3-氧雜-1-戊烯)。
合適的(b)類型的烯鍵式不飽和氟化單體的非限制性實例是:
c2-c8氟烯烴,諸如四氟乙烯、五氟丙烯、六氟丙烯、以及六氟異丁烯;偏二氟乙烯;
c2-c8氯代-和/或溴代-和/或碘代-氟烯烴,諸如氯代三氟乙烯和溴代三氟乙烯;具有式cf2=cforf1的氟烷基乙烯基醚,其中rf1是c1-c6氟烷基,例如-cf3、-c2f5、-c3f7;具有式cf2=cforo1的氟-氧烷基乙烯基醚,其中ro1是具有一個或多個醚基的c1-c12氟-氧烷基,例如全氟-2-丙氧基-丙基;具有式cf2=cfocf2orf2的氟烷基-甲氧基-乙烯基醚,其中rf2是c1-c6氟烷基,例如-cf3,-c2f5,-c3f7,或具有一個或多個醚基的c1-c6氟氧烷基,像-c2f5-o-cf3;具有下式的氟間二氧雜環戊烯:
其中rf3、rf4、rf5、rf6中的每一個,彼此相同或不同,獨立地是氟原子、c1-c6氟(鹵)氟烷基,任選地包含一個或多個氧原子,例如-cf3、-c2f5、-c3f7、-ocf3、-ocf2cf2ocf3。
優選地,單體(b)在以下各項中選擇:
c3-c8氟烯烴,優選四氟乙烯和/或六氟丙烯;
氯代-和/或溴代-和/或碘代-c2-c6氟烯烴,像氯代三氟乙烯和/或溴代三氟乙烯;
具有式cf2=cforf1的氟烷基乙烯基醚,其中rf1是c1-c6氟烷基,例如-cf3、-c2f5、-c3f7;
具有式cf2=cforo1的氟-氧烷基乙烯基醚,其中ro1是具有一個或多個醚基的c1-c12氟氧烷基,像全氟-2-丙氧基-丙基。
最優選地,單體(b)是四氟乙烯。
本發明的方法中的該組合物可以包含呈中性形式的至少一種氟化聚合物,其中表述“中性形式”指示在-so2x官能團中x是x’并且x’選自由f、cl、br、i組成的組中。優選地,x’選自f或cl。更優選地,x’是f。
可替代地,該組合物可以包含呈離子(酸或成鹽的)形式的至少一種氟化聚合物,其中表述“離子形式”指示在-so2x官能團中,x是oz并且z選自由h、堿金屬、nh4組成的組中。
為避免疑義,術語“堿金屬”在此旨在表示下面的金屬:li、na、k、rb、cs。優選地,該堿金屬選自li、na、k。
包含-so3z官能團的氟化聚合物典型地通過本領域中已知的方法由包含-so2x’官能團、優選-so2f官能團的氟化聚合物制備。該氟化聚合物可以通過用強堿(例如naoh、koh)處理相應的包含-so2x’官能團、典型地-so2f官能團的聚合物以其成鹽的形式獲得,即其中z是選自由nh4和堿金屬組成的組的陽離子。該氟化聚合物可以通過用濃酸溶液處理相應的成鹽形式的聚合物以其酸形式獲得,即其中z是h。
包含-so2x’官能團的氟化聚合物可以通過本領域中已知的任何聚合方法制備。用于此類聚合物的制備的適合的方法是例如在us4940525a(陶氏化學公司(thedowchemicalcompany))7/10/1990、ep1323751a(蘇威蘇萊克斯公司(solvaysolexisspa))7/2/2003和ep1172382a(蘇威蘇萊克斯公司)11/16/2002中描述的那些。
有待用于本發明的方法中的氟化聚合物還可以通過磺酸氟化聚合物的交聯獲得,該交聯涉及聚合物的骨架。這些氟聚合物可以是任何類型,還適合用作電化學電池中的離子傳導膜以及過濾膜兩者。
之前已經披露了利用交聯來改進由包含磺酸官能團的氟化聚合物制造的催化劑的物理抗性。例如,ep1238999a(蘇威蘇萊克斯公司)9/11/2002和ep1239000a(蘇威蘇萊克斯公司)9/11/2002披露了包含可交聯的磺酸氟化聚合物的親水性膜,這些聚合物包含:衍生自四氟乙烯的單體單元、包含磺酰基團-so2f的氟化的單體單元,以及衍生自具有式(i)的雙-烯烴的按摩爾計從0.01%至5%的單體單元:r1r2c=ch-(cf2)m-ch=cr5r6
(其中m=2-10,r1、r2、r5、r6,彼此相同或不同,是h或c1-c5烷基)。
適合用于本發明的方法中的氟化聚合物的非限制性實例是商業產品
該催化劑載體可以通過磺酸氟化聚合物的交聯得到,該交聯涉及聚合物的骨架。由此獲得的聚合物適合用作電化學電池中的離子傳導膜以及過濾膜兩者。之前也已經描述了氟化聚合物的交聯也可能涉及磺酸官能團的磺酰基氟化物官能團前體。
根據本發明的方法中的雙功能催化劑的可用性允許在一鍋法中或者依次或以合作方式有效地進行兩種反應并使用單一催化材料:酸催化的反應(脫水、酯化、異構化、水解)和金屬催化的還原。本發明的方法的優點包括最終產物的高純度、由多步驟制備所要求的操作的較低數目、任何可溶性酸性添加劑的避免、所要求的低能量輸入(從而減少了時間和成本(初始的和操作的))、以及方法復雜性的降低(不需要中間體的純化和/或回收)。
目前,許多現有技術方法用均相催化劑進行。非均相催化的使用允許更容易的催化劑分離和產物純化。
在根據本發明的方法中,根據任何順序,該氫化和酸催化的脫水/異構化可以同時或依次進行,即該氫化可以在該脫水/異構化之前或之后,取決于該底物的特定反應性和所涉及的中間體化學物種的特定反應性,以最終產生氫化的且脫水的/異構化的化合物。
使用根據本發明的基于氟聚合物的催化劑意味著在本發明的方法過程中離聚物結構與具有與離聚物的離子官能團的親和力的水性或極性液體的接觸。總體上,存在于該鏈中的離子基團的量越大,就本發明的方法中的催化劑活性而言的效率越好。
從這個觀點來看,重要的參數是離聚物的當量重量。氟化聚合物(p)中的-so3m官能團的量(就酸基團-so3h而言測量的)與聚合物的所謂的“當量重量(ew)”相關,該當量重量是每摩爾的酸官能團的氟化聚合物的克數(指示為“eq”)。
該聚合物的當量重量(ew)可以通過ir分析來確定。當量重量越低,該鏈中存在的磺酸基的百分比越高。因此,具有低當量重量的離聚物是令人希望的,因為它們給出較高的催化效率。該氟化聚合物(p)典型地具有500g/eq或以上、優選地不小于450g/eq或以上、更優選地380g/eq或以上的當量重量。該當量重量典型地不超過1600g/eq,優選地其不超過1200g/eq,更優選地其不超過900或1000g/eq。
該氟化聚合物(p)具有在室溫下每1g干燥聚合物至少0.5ml所吸收的溶劑(典型地水)的典型的重量溶脹能力,其或者以在60-500mm的范圍內的晶粒尺寸的粉末的形式(如經由本領域技術人員已知的篩分技術確定的,例如astm1921-12或作為具有在1-50mm的范圍內的孔徑的大網狀泡沫可獲得的。該氟化聚合物(p)允許錨定過渡金屬顆粒,相對于so3h基團的總摩爾數,其量是從5至50摩爾%、優選10至40摩爾%、更優選20至30摩爾%,取決于包括金屬的化合價的因素。
在本發明的上下文中,表述“至少一種”當指的是“氟化聚合物”和/或“金屬顆粒”時旨在表示一種或多于一種聚合物和/或一種或多種類型的金屬顆粒。聚合物和/或金屬顆粒的混合物可以有利地用于本發明的目的。
術語“過渡金屬”指示金屬化學元素,其原子具有不完全的d亞層,或者其可以產生具有不完全d亞層的陽離子,即周期表的d-區中的元素,其包括周期表上的第3族至第12族,即第一(3d)過渡系列(sc-zn)、第二(4d)過渡系列(y-cd)和第三(5d)過渡系列(lu-hg)。
優選地,在本發明的方法中聚合物載體(p)上的金屬顆粒包含至少一種或者由至少一種非電荷或離子形式的金屬組成,所述金屬選自釕、銠、鉑、鈀、銅、鎳、銥、鈷、鋨、錸、鐵及其組合。更優選地,該金屬是釕、鉑、鈀或銠。
在本發明的上下文中,術語“顆粒”旨在表示,從幾何學觀點來看,具有明確的三維體積和形狀的材料塊體,其特征為三個維度,其中所述維度中沒有一個比其余兩個另外維度超出大于200%。顆粒總體上不是等維度的,即在一個方向上比其他方向上更長。
顆粒的形狀可以值得注意地就球形度φs而言表示,其獨立于粒徑。顆粒的球形度是具有與該顆粒相等體積的球體的表面-體積比與該顆粒的表面-體積比的比率。對于直徑dp的球形顆粒,φs=1;對于非球形顆粒,球形度被定義為
φs=(6.vp)/(dp.sp)
其中:
dp是顆粒的當量直徑;
sp是一個顆粒的表面積;
vp是一個顆粒的體積。
當量直徑被定義為相等體積的球體的直徑。基于篩分析或微觀分析,dp通常被以為是標稱尺寸。表面積是從吸附測量值或從顆粒床的壓降發現的。聚合物(p)的顆粒具有優選至少0.6、更優選至少0.65、甚至更優選至少0.7的球形度φs。使用具有從0.7至0.95的φs的顆粒已經獲得了良好的結果。
如在此使用的,術語“粒徑”指的是如通過本領域技術人員已知的一般方法如篩分析或微觀分析確定的金屬顆粒的當量直徑(dp)。
本發明的方法中的金屬顆粒的粒徑低于50nm(“納米顆粒”)、優選低于20nm、更優選低于10nm、甚至更優選低于5nm。在特別優選的實施例中,根據本發明的催化劑中的至少90%的金屬顆粒具有低于9nm的尺寸。粒徑是根據本領域技術人員常規使用的且已知的方法測定的,如經由透射電子顯微鏡、x射線衍射、化學吸附、小角x射線散射。
發現,根據本發明的用于獲得產物(ii)的含有至少一個c=c或c=o部分的底物(i)的一鍋法氫化和酸催化異構化或脫水可以有利地在相對低的溫度和氫氣氣體(h2)的中等壓力下進行。因為可以使用相對溫和的條件,本發明的整體方法是特別成本效益的并且使用本領域技術人員已知的常規程序可擴展到工業生產。
根據本發明的方法可以在1atm至50atm的氫氣壓力下在40℃與200℃之間進行。
優選地,在根據本發明的方法中,在氫氣的存在下使該底物(i)與催化劑(iii)反應的步驟是在從50℃至150℃的溫度下、更優選在低于100℃的溫度下、甚至更優選在室溫下并且在低于50atm、更優選低于30atm并且甚至更優選低于10atm的氫氣壓力下進行的。在氫氣的存在下與該底物(i)的反應中存在的催化劑(iii)的量可以根據正在使用的過渡金屬,根據該底物的化學結構,根據反應條件如溶劑的性質、氫氣的壓力、溫度等而變化,如有機合成領域中的技術人員已知的。優選地,相對于底物(i)的重量,催化劑(iii)的量是以重量計從1:1至1:200,對應于基于催化劑(iii)中的過渡金屬的0.05至10%重量負載量的1:100至1:10000的催化劑/底物摩爾比范圍。
發現根據本發明的方法可以在相對于該底物的摩爾量的小于1mol%的過渡金屬的存在下有效地進行,而在現有技術的方法中總體上要求至少5mol%的金屬。這在通過金屬(像鉑和鈀)催化的方法中是特別有利的,其中有待用作催化劑的貴金屬的量是整個方法成本的主要組成。
優選地,催化劑(iii)中的過渡金屬的量是以重量計小于3%、更優選以重量計小于2%、最優選小于1%/該催化劑的總重量。
有利地,根據本發明的方法的在催化劑(iii)的存在下使底物(i)與氫氣反應的步驟可以在水的存在下進行。涉及水性溶劑的化學轉化是比唯一地在有機溶劑中進行的方法更環保并且總體上更成本效益的。
優選地,在根據本發明的方法中,在催化劑(iii)的存在下使底物(i)與氫氣反應的步驟中的溶劑包含水,更優選以相對于該溶劑的總重量至少50%重量、甚至更優選相對于該溶劑的重量至少80%或90%重量。優選地,根據本發明的方法在水和水可混溶有機溶劑的混合物中進行,包括但不限于,水-甲醇、水-乙醇、水-1,4-二噁烷和水-丙酮。
根據本發明的用于獲得產物(ii)的含有至少一個c=c或c=o部分的底物(i)的一鍋法氫化和酸催化異構化或脫水可以有利地用于生產若干種高級中間體和復雜化學品。
在本發明的優選的實施例中,通過在氫氣的存在下與催化劑(iii)反應將乙酰丙酸(ia)轉化為γ-戊內酯(iia)(方案1)。
方案1
發現(iia)可以根據本發明的方法以良好的產率(>48%)和優異的選擇性還在低溫(50℃)下制備。相比之下,在類似條件下在50℃下處理(ia)時僅獲得29%的(iia),除了使用包含負載在非氟化樹脂(
此外,時常作為根據現有技術的方法制備的(iia)中的雜質發現的中間體γ-羥基戊酸(hva)不存在于根據本發明的方法獲得的(iia)中(hva的含量低于gc分析的檢測限)。
優選地,在根據本發明的(ia)至(iia)的轉化中,該催化劑(iii)包含釕、銠、鈀或鉑,更優選釕。
優選地,在根據本發明的(ia)至(iia)的轉化中,該催化劑(iii)具有從1100至500g/eq、更優選地1000至600g/eq的ew。
優選地,在根據本發明的(ia)至(iia)的轉化中,該溶劑由水組成或包含相對于該溶劑的總重量以重量計至少80%、更優選至少90%的水。
在本發明的另一個優選的實施例中,通過在氫氣的存在下與催化劑(iii)反應將半纖維素或纖維素(ib)轉化為山梨醇(iib)(方案2)。
方案2
優選地,在根據本發明的(ib)至(iib)的轉化中,該催化劑(iii)包含釕、銠、鈀或鉑,更優選釕。
山梨醇(iib)作為低卡路里甜味劑、保濕劑、瀉藥并且作為重要物質如表面活性劑和維生素c的前體廣泛用于食品和制藥工業中。它還是用于制備若干種化學品(包括乙二醇、丙二醇、甘油和異山梨醇)的平臺分子。目前,山梨醇通過淀粉的酶水解和催化氫化以商業規模生產。有利地,本發明的方法提供了從半纖維素或直接從纖維素(自然界中最豐富的有機化合物之一)開始的山梨醇的路線,該纖維素是不可食用的并且可以容易地從低成本材料以及從來自若干食品/工業過程的廢物獲得。
在本發明的另一個優選的實施例中,通過在氫氣的存在下與催化劑(iii)反應直接地將木糖(ic)轉化成乙酰丙酸或其酯(iic)(方案3)。
方案3
乙酰丙酸(ia)及其酯(iic)是用于制備“綠色”溶劑以及多種結構單元和生物燃料的重要起始材料。它們目前通過在高溫下用濃hcl或硫酸處理蔗糖大規模地獲得。根據本發明的方法在溫和條件下并且以高產率提供了從(ic)開始的(iic)和(ia)的一鍋法路線。可替代地,(iic)和(ia)可以根據本發明的方法使用糠醛(通過不需要被離析的中間體糠醇)來制備。
在本發明的另一個優選的實施例中,通過在氫氣的存在下與催化劑(iii)反應將γ-戊內酯(iia)轉化為戊酸(iid)(方案4)。
方案4
iid(戊酸(valericacid)或戊酸(pentanoicacid))是用于若干種方法的重要高級中間體。值得注意地,通過酯化,它提供了可用作可再生燃料并且可用于食品和香料工業中的戊酸烷基酯。
在本發明的另一個優選的實施例中,通過在氫氣的存在下與催化劑(iii)反應將甘油(ie)轉化為1,2-丙二醇(iie)(方案5)。
方案5
丙二醇(1,2-丙二醇,iie)是在150℃-200℃下在強酸存在下由環氧丙烷工業生產的。此種方法的最終產物含有20%的丙二醇、1.5%的二丙二醇和少量的其他二醇。根據本發明的方法,可以從非常常見的起始原料甘油(ie)以高產率和純度獲得(iie),該甘油是大規模地從脂肪和植物油中發現的甘油三酯或作為來自從植物油生產生物柴油的副產物獲得的。
在本發明的另一個優選的實施例中,通過在氫氣的存在下與催化劑(iii)反應將香茅醛(if)轉化成薄荷醇(iif)(方案6)。
方案6
薄荷醇是最需要的薄荷產物,廣泛用于調味、食品、保健和制藥工業中。合成的薄荷醇(特別地天然存在的(-)異構體)通常是通過tagasako方法獲得的,該方法基于從月桂烯開始的五個步驟。本發明的方法可以應用于香茅醛,或者外消旋的或同分異構地富集的(r或s旋光異構體),以在一鍋法中在非手性催化劑存在下以高純度獲得外消旋(+)-或(-)-薄荷醇。
在本發明的另一個優選的實施例中,通過在氫氣的存在下與催化劑(iii)反應將丙酮(ig)轉化成甲基異丁基酮(iig)(方案7)。
方案7
甲基異丁基酮(iig,mibk)是從丙酮獲得的最重要的化合物,每年具有數百萬千克的全球生產,并且廣泛用于漆料、樹脂和涂料以及作為若干方法中的溶劑用于化學工業中。mibk的商業生產涉及三個步驟,并且該方法的缺點是使用分開的反應單元、高溫和大量的廢物。本發明的方法提供了一種可行的一鍋法路線,該路線用于使用較低的溫度以高產率和純度從丙酮生產mibk,并且相對于商業方法產生較少的副產物。
在本發明的另一個優選的實施例中,通過在氫氣的存在下與該催化劑(iii)反應將葡萄糖(ih)轉化成1,6-己二醇(ii’h)或2,5-二甲基呋喃(iih)(方案8)。
方案8
本發明的方法提供了1,6-己二醇的途徑,該1,6-己二醇主要用于從葡萄糖開始生產尼龍、聚酯和聚氨酯,作為使用己二酸作為起始原料的當前商業方法的替代方案。
2,5-二甲基呋喃作為汽油摻和劑或辛烷值增進劑用于燃料工業中并且是用于制造聚氨酯泡沫或聚酯的起始材料。值得注意地,根據本發明從葡萄糖合成2,5-二甲基呋喃是有利的,因為該起始材料是負擔得起的、容易從可再生來源可獲得的并且容易大規模地可獲得的。
該催化劑(iii)可以根據本領域技術人員已知的標準程序來制備。用于制備可用于本發明的方法中的催化劑的程序的非限制性實例披露于us2008/0003479a(建國大學(konkukuniversity))1/3/2008和wo2013/162499a(聯合技術公司(unitedtechnologiescorporation))10/31/2013中。
在實施例中,本發明提供了一種固體組合物,該固體組合物包含負載在包含至少一種帶有-so2x官能團的氟化聚合物(p)的材料上的粒徑低于50nm的過渡金屬顆粒,其中x選自x'和om,x'選自由f、cl、br和i組成的組;并且m選自由h、堿金屬和nh4組成的組,并且(p)包含衍生自四氟乙烯(tfe)和直鏈(即非支鏈的)磺酰基氟化物乙烯基醚(sfve)的重復單元。
優選地,(p)包含衍生自具有式cf2=cf-o-(cf2)n-so2f的直鏈sfve的單元,其中n=2、3或4,更優選地其中n=2。
發現,根據本發明的組合物可以有效地用作底物的一鍋法氫化和酸催化異構化或脫水中的催化劑。使用本發明的組合物作為催化劑,相對于該底物的摩爾量,小于1mol%的過渡金屬足以在溫和條件下以良好至優異的選擇性完全轉化,而至少5mol%的金屬和更苛刻的條件總體上在現有技術的方法中被要求,對該方法的整體成本具有顯著的影響。
優選地,在根據本發明的固體組合物中,這些過渡金屬顆粒的粒徑低于20nm、更優選低于10nm、甚至更優選低于5nm。在特別優選的實施例中,根據本發明的組合物中的至少90%的金屬顆粒的尺寸是低于9nm。
優選地,在所述固體組合物中,該載體(p)上的金屬顆粒包含至少一種或者由至少一種非電荷或離子形式的金屬組成,所述金屬選自釕、銠、鉑、鈀、銅、鎳、銥、鈷、鋨、錸、鐵及其組合,優選地其中該金屬是釕、鉑、鈀或銠。
優選地,在所述固體組合物中,該聚合物(p)具有每摩爾的-so3h官能團以氟化聚合物的克數計的500g/eq或以上、優選地450g/eq或以上、更優選地380g/eq或以上的當量重量。
若通過援引方式并入本申請的任何專利、專利申請以及公開物的披露內容與本申請的描述相沖突到了可能導致術語不清楚的程度,則本說明應該優先。
提供以下實例來說明本發明的典型實施例并且這些實例不旨在限制本發明的范圍。
esem(環境掃描電子顯微鏡)實驗在feiquanta200顯微鏡上進行,該顯微鏡在25kev的加速電壓下操作并配備有edax能量色散x射線光譜儀(eds)。在120kev的m12philips儀器上進行tem(透射電子顯微鏡)分析。
通過使用varian720es儀器的電感耦合等離子體原子發射光譜法(icp-oes)確定這些催化劑中的金屬負載量和溶液中潛在地瀝濾的金屬的量。
在所有實例中,沒有觀察到金屬到該反應溶液中的瀝濾。
在配備有火焰離子化檢測器(fid)和30m(0.25mmid,0.25μmft)vf-waxms柱的shimadzugc2010色譜儀上進行gc分析。
在配備有相同柱的shimadzuqp5000裝置上進行gc-ms分析。
在分批條件下的反應使用配備有磁力攪拌器、
實例i.含ru催化劑的制備
在典型的程序中,將干燥聚合物材料
然后將固體nabh4過量地(30:1)并且在氮氣下加入到含有以上獲得的金屬化的
從icp-oes分析中測量了1.16wt%的釕負載量,對應于80.0%的金屬吸收。
實例ii.含rh催化劑的制備
在氮氣下經由注射器將[rh(nbd)2]bf4(32.9mg,0.088mmol,比率so3h/rh=10.5)在thf(17.0ml)中的脫氣橙色溶液加入到thf(17.0ml)中的脫氣懸浮液(600mg)中。將該混合物使用軌道攪拌器在室溫下攪拌24h。在高真空下干燥過夜之前,將所得固體材料傾析出,用thf(3×25ml)和二乙醚(20ml)洗滌。在氮氣下儲存由此獲得的含金屬的
然后在氮氣下將固體nabh4(100mg)加入到該含金屬的樹脂在水(15ml)中的脫氣懸浮液中。使該混合物在軌道攪拌下反應24h。在該時間后,將該固體傾析出,用脫氣水(4×25ml)洗滌,并在高真空下在50℃干燥過夜。在氮氣下儲存作為黑色粉末獲得的所得產物。
從icp-oes分析中測量了1.41wt%的銠負載量,對應于95.3%的金屬吸收。
實例iii.含pd催化劑的制備
在典型的程序中,在氮氣下經由注射器將pd(no3)2(23.4mg,0.088mmol,比率so3h/rh=10.5)在水(17.0ml)中的脫氣褐色溶液加入到
從icp-oes分析中測量了1.33wt%的鈀負載量,對應于86.9%的金屬吸收。
實例iv.含pt催化劑的制備
將干燥聚合物材料
然后將固體nabh4過量地(30:1)并且在氮氣下加入到含有金屬化的
從icp-oes分析中測量了2.53wt%的鉑負載量,對應于91.7%的金屬吸收。
催化反應
實例v.將乙酰丙酸(ia)直接催化轉化為γ-戊內酯(iia)。
在氮氣下將實例i的負載型催化劑(60mg,1.16wt%ru,0.007mmol的釕)放置于不含金屬的高壓釜中。將乙酰丙酸(0.43m,2.15mmol)在去離子水(5.0ml)中的脫氣溶液在氮氣下經由特氟龍管轉移到該高壓釜中。用三次循環加壓/減壓將氮氣替換為氫氣。最后將該高壓釜裝入5巴壓力的氫氣,使用機械攪拌器以150rpm攪拌并使用油浴加熱到高達70℃。在4小時之后,將該反應器冷卻至室溫,減壓,并使用氣密注射器完全去除該溶液。將該溶液的樣品用于gc和hplc(產物產率)、gc-ms和nmr(產物鑒定)和icp-oes分析(金屬瀝濾)。檢測到的唯一產物是γ-戊內酯,具有79.8%的產率,對應于62h-1的奈奎斯特頻率(轉化的底物的摩爾數/金屬摩爾數xh)。沒有觀察到痕量的γ-羥基戊酸(hva)。
實例vi將乙酰丙酸(ia)直接催化轉化為γ-戊內酯(iia)。
重復實例v的程序,唯一的區別是使用油浴將高壓釜加熱至50℃而不是70℃。在4h之后獲得的唯一產物是γ-戊內酯,具有48.5%的產率,對應于38h-1的奈奎斯特頻率。沒有觀察到痕量的γ-羥基戊酸(hva)。
對比實例i.使用非氟化催化劑將乙酰丙酸(ia)催化轉化為γ-戊內酯(iia)
重復實例vi的程序,使用負載在非氟化催化劑上的釕(負載在
實例vii.將(r)-(+)-香茅醛(1f)直接催化轉化為薄荷醇(iif)
在氮氣下將實例iii的負載型催化劑(60mg,1.33wt%pd,0.007mmol的鈀)放置于不含金屬的高壓釜中。然后在氮氣下加入純凈的(r)-(+)-香茅醛(133.0mg,0.86mmol)。將脫氣且去離子的水(9.0ml)在氮氣下經由特氟龍管轉移到該高壓釜中。經由三次循環加壓/減壓將氮氣替換為氫氣。最后將該高壓釜裝入10巴壓力的氫氣,使用機械攪拌器以150rpm攪拌并使用油浴加熱到高達80℃。在48小時之后,將該反應器冷卻至室溫,減壓,并使用氣密注射器完全去除該溶液。將由此獲得的溶液的樣品用于gc和hplc(產物產率)、gc-ms和nmr(產物鑒定)和icp-oes分析(沒有觀察到金屬瀝濾)。檢測到的唯一產物是(-)-薄荷醇,具有100.0%的起始香茅醛的轉化率。沒有觀察到痕量的異胡薄荷醇或任何其他副產物。
- 還沒有人留言評論。精彩留言會獲得點贊!