聚合物鏈修飾的硅膠基質親水作用色譜固定相及其制備和應用的制作方法
本發明涉及一種親水作用液相色譜固定相及其制備方法,以及其在分離分析、糖蛋白質組學中的應用。
背景技術:
高效液相色譜(HPLC)技術自20世紀70年代以來在儀器系統、固定相、分離理論等方面都得到了巨大的發展。由于其高效和快速的特點,該技術被廣泛應用于不同的研究領域,如制藥、生命科學、環境分析和食品安全等(Fekete,S et al,TrAC.2014,63,2-13)。固定相作為色譜技術的核心,從根本上決定了色譜的分離選擇性和分離效率。近來年,隨著蛋白質組學、代謝組學、藥物分析等領域的不斷發展,強極性物質的分離備受各個領域的關注。親水作用色譜對強極性和親水化合物具有很好的保留和分離選擇性,且親水模式使用的流動相體系相對簡單,分離選擇性與反相色譜具有很好的正交性,因此越來越受到關注和重視。
目前,已經有多種類型的親水作用色譜固定相,如酰胺型(R.El-Debs et al,JCA.2014,1326,89-95)、多元醇羥基型(Franc,ois M et al,JCA.2010,1217,7528-7538)、兩性離子型(Hongdeng Q et al,CC,2013,49,2454-2456)等并且已經商品化。但上述固定相仍然存在親水性不足、分離選擇性不夠、制備復雜等問題。此外,值得注意的,使用不同方法制備的親水作用固定相,其極性功能基團的連接方式和空間排布不同,得到的固定相的表面結構和分離性能存在很大差別(Strege,M.A.et al,AC.1998,70,2439-2445)。
技術實現要素:
為了解決上述問題,本發明的目的在于提供一種聚合物鏈修飾的硅膠基質親水作用色譜固定相,該固定相通過硫醇-烯點擊共聚反應在硅膠表面鍵合由兩種不同單體形成的樹枝狀聚合物鏈,該固定相表面富含羥基和酰胺鍵多種親水性功能基團,具有很好的親水性,在分離分析和糖蛋白質組學等領域有較好的實用價值和應用前景。
本發明所采用的技術方案為:
一種聚合物鏈修飾的硅膠基質親水作用色譜固定相,以硅膠顆粒為基質,第一步進行硅烷化修飾上巰基,然后通過硫醇-烯的點擊共聚反應在硅膠表面接枝含有羥基的烯類功能單體和甲叉雙丙烯酰胺功能單體,形成表面具有三維樹枝狀聚合物鏈且富含羥基和酰胺親水性功能基團的親水作用色譜固定相。
所述親水作用固定相表面具有三維樹枝狀聚合物鏈,固定相含有羥基 和酰胺兩種親水作用功能基團。
所述固定相顆粒為單分散的微球顆粒,固定相顆粒粒徑在500nm-10μm之間。
所述基質為核/殼結構或全多孔結構的硅膠顆粒;核/殼結構硅膠顆粒由700nm-3μm直徑的實心硅膠核和殼層厚度為150nm-500nm的多孔硅膠外層組成,孔徑為3nm-20nm;全多孔結構硅膠顆粒粒徑為500nm-10μm,孔徑為5nm-100nm。
所述功能單體為兩種,一種是含有羥基的烯類功能單體,其結構為:
其中Y代表-CONH-或者-OOC-;Z代表-CH2OH;m的值為1-10的正整數,n的值為1-3的正整數;
另一種為甲叉雙丙烯酰胺。
本發明的另一個目的在于提供一種聚合物鏈修飾的硅膠基質親水作用色譜固定相的制備方法。該親水作用色譜固定相不僅可以實現高選擇性分離強極性化合物和親水性物質,而且制備方法簡單普適,容易實現。
具體步驟為:
a)硅烷化修飾硅膠顆粒的過程:將活化后的硅膠顆粒分散在甲苯中,超聲,加入帶有硫醇基團的硅烷化試劑,通氮氣5-30分鐘,攪拌,加熱回流;停止反應后,冷卻至室溫,離心,棄上清液,之后將得到的固體顆粒依次使用甲苯、甲醇和丙酮抽濾洗滌,在真空干燥箱中至恒重;
b)聚合物鏈鍵合相的制備:將得到的硅烷化修飾硅膠顆粒、功能單體分散在反應溶劑中,超聲,通氮氣5-30分鐘,加入引發劑,機械攪拌,在50-100℃下反應,停止反應,冷卻至室溫,離心,去除上清液,抽濾,并依次使用反應溶劑、水、甲醇洗滌微球,重復洗滌,在真空干燥箱中至恒重,既得聚合物修飾的硅膠液相色譜固定相。
步驟a)中攪拌回流時間為6-30h,加熱溫度為100-130℃,離心速度2500~10000rad/min,離心時間3-5分鐘,重復抽濾洗滌次數為2~5遍,室溫下真空干燥,時間為6-30小時;
步驟b)中反應時間為4-48小時,反應過程中為均勻緩慢加熱至所需溫度;離心速度為3000~10000rad/min,重復洗滌次數為2~5遍,室溫下真空干燥6-30小時。
步驟a)中硅烷化試劑為含有巰基的硅烷化試劑。
所用的硅烷化試劑具有如下結構:
其中X代表甲氧基或者乙氧基,m的值為1-4的正整數;
其中硅膠顆粒與硅烷化試劑的加入量質量比為1:0.05至1:1。
硅膠顆粒占反應體系總質量的0.05~0.1wt%,硅烷化試劑的總摩爾數濃度為0.04~1mol/L,余量為反應溶劑。
步驟b)中所述兩種功能單體的摩爾比為0.1:1-1:0.1;
步驟b)中甲叉雙丙烯酰胺和含有羥基的烯類功能單體在反應溶劑體系中的總摩爾數濃度為0.05-0.4mol/L;硅膠顆粒加入質量與兩種單體總質量比為1:0.1至1:10;
步驟b)所用的引發劑為偶氮類引發劑,包括偶氮二異丁腈、偶氮二異庚腈、偶氮二異丁脒鹽酸鹽、偶氮二異丁咪唑啉鹽酸鹽中的一種或兩種及以上組合,加入總量占兩種功能單體總質量的0.5~10%;
步驟b)所用的溶劑為甲醇、乙醇、乙腈、甲苯、丙醇、N,N-二甲基甲酰胺、水中一種或二種及以上混合溶液。
本發明所述聚合物鏈修飾的硅膠基質親水作用色譜固定相能夠應用于極性物質和親水性物質的色譜分離或糖肽、糖蛋白的富集和分離。
所述親水作用固定相能夠應用于核苷、堿基、氨基酸、酸性小分子、堿性小分子、肽段和蛋白質中的一種或兩種以上的分離,能夠應用于糖肽和糖蛋白的富集;分離和富集所適用的pH范圍為2-8,流動相為體積濃度為50%-98%的乙腈,緩沖鹽和水,其中,緩沖鹽種類為乙酸銨,甲酸銨,磷酸二氫鉀,磷酸二氫鈉,緩沖鹽濃度為0-120mM。
本發明具有如下優點:
本發明在材料表面通過硫醇-烯點擊共聚的方法引入了特殊空間結構的羥基、酰胺基等親水性功能基團,克服了傳統后修飾方法步驟繁瑣、反應效率低的缺點,制備的硅膠顆粒固定相表面具有三維結構的聚合物鏈,該結構可以提高材料的親水性。
該制備方法操作簡單,反應效率高,反應條件溫和;固定相顆粒分散性好,粒徑均勻,粒徑分布窄;表面三維聚合物鏈狀結構,富含多種親水性基團,所制備的固定相親水性好,分離選擇性好;所制備固定相應用范圍廣,可廣泛應用于強極性和親水性樣品分離分析和糖蛋白質組學。
附圖說明
圖1為實施例1中制備的聚合物微球的紅外測試結果圖;
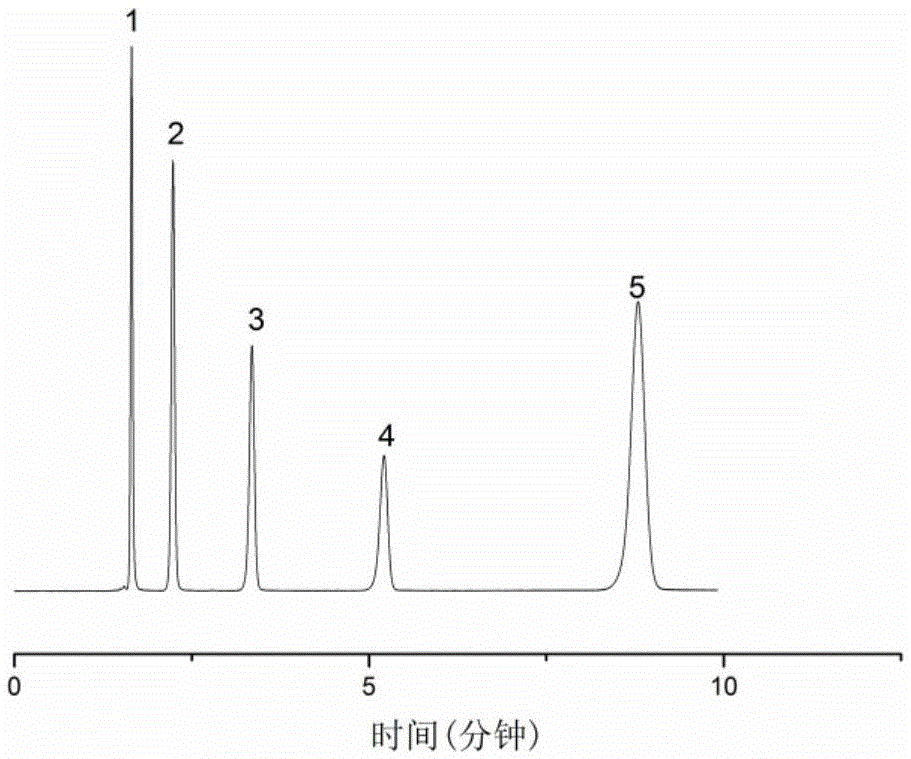
圖2為親水作用色譜固定相用于核苷的分離色譜圖;
圖3為親水作用色譜固定相用于有機酸的分離色譜圖;
圖4為親水作用色譜固定相用于糖肽富集效果圖。
具體實施方式
實施例1
1、巰基化硅球的制備:在250mL的圓底燒瓶中,加入120mL甲苯,加入5g粒徑為5μm的硅膠顆粒,2ml的3-巰丙基三乙氧基硅烷,超聲2分鐘分散均勻,在燒瓶上接冷凝管,機械攪拌,保持300rad/min速度,通氮氣10分鐘。反應裝置放在油浴鍋中,110℃加熱回流13h,停止反應,冷卻至室溫。高速離心機3000rad/min的速度離心,去除上清液,依次使用甲苯、丙酮、甲醇、丙酮抽濾洗滌,重復抽濾洗滌3遍洗三遍,50℃真空干燥箱內真空干燥24小時,得到修飾了巰基的硅膠顆粒。
2、硅膠表面親水聚合物鏈的制備:在250mL的圓底燒瓶中,加入反應溶劑水-乙醇混合溶液(體積比為2:1)100mL,再加入2000mg修飾硅烷化的硅膠顆粒、1000mg N-[三(羥甲基)甲基]丙烯酰胺(THMA)、900mg甲叉雙丙烯酰胺酸、100mg偶氮二異丁腈(AIBN)超聲1分鐘,使得加入的試劑以及顆粒溶解形成均勻分散在溶劑中,之后通氮氣15分鐘,機械攪拌,保持300rad/min速度。反應裝置放在油浴鍋中均勻加熱,在30min內升溫至75℃。維持75℃條件下反應12小時,停止反應,冷卻至室溫,得到微球顆粒silica@(MBAAm-co-THMA)之后使用高速離心機用10000rad/min的速度離心,去除上清液,加入反應溶液洗三遍,50℃真空干燥箱內真空干燥24小時。得到固定相顆粒silica@(MBAAm-co-THMA)。
3、固定相顆粒的表征
經過紅外分析測試,結果如圖1所示。固定相顆粒在1382cm-1表現出與亞甲基相連的羥基的特征峰,在1531cm-1和1650cm-1處表現出酰胺的特征吸收峰,在2956cm-1和2882cm-1表現出烷烴的碳-氫鍵的吸收峰,證明了固定相的結構,說明了這種方法制備固定相的可行性。
實施例2
把實施例1所制備的固定相顆粒填裝于4.6mm*150mm的不銹鋼高效液相色譜柱中,制備的色譜柱用于在親水模式下分離核苷和堿基的混合物。分離條件為:乙腈/水(體積比為81比19)作為流動相,流速1ml/min,柱溫為25℃,分離色譜圖如圖2所示,圖中1,2,3,4,5分別為胸苷,尿苷,胞嘧啶核苷,胞嘧啶,鳥苷。分離結果顯示,在本發明提供的親水作用色譜柱上,核苷和堿基得到了很好的基線分離,表明該固定相具有典型的親水作用特征和良好的分離選擇性。
實施例3
使用實施例2所制備的親水作用色譜柱分離有機酸性化合物。分離條件為:以88%的乙腈,2%的醋酸銨(濃度為100mM),和10%的水作為流動相,流速1ml/min,柱溫為25℃。分離色譜圖如圖3所示,圖中1,2,3,4,5分別為苯酚,對苯甲酚,3,5-二硝基苯甲酸,苯甲酸,4-羥基苯甲酸。分離色譜圖顯示這幾種有機酸在本發明提供的色譜柱上具有很好的保留,得到了良好的基線分離。
實施例4
1、把實施例1所制備的固定相顆粒填裝于4.6mm*10mm的不銹鋼色譜柱中,將制備的色譜柱用于糖肽富集。將糖蛋白(免疫球蛋白G,IgG)經過trypsin酶解,除鹽凍干,并溶解于上樣溶液中,從而制得濃度為500ng/μL的蛋白混合溶液。上樣條件為85%的乙腈,15%的水和0.1%的三氟乙酸,上樣量為100μL,洗脫條件為50%的乙腈,50%的水和0.1%的三氟乙酸.收集洗脫液冷凍濃縮得到富集產物。將酶解產物原液和富集產物進行MALDI-TOF MS鑒定。
2、將1μL待分析物與1μL DHB基質(20mg/mL 2,5-二羥基苯甲酸溶于含1%三氟乙酸的60%乙腈溶液)依次點于MALDI靶板上,待樣品點干燥后進行質譜鑒定。
如圖4所示,a圖為未經過富集處理的IgG蛋白酶解產物原溶液,b圖為富集產物。如圖4a所示,在IgG蛋白酶解產物溶液中沒有鑒定到IgG的糖肽,經富集之后(圖4b)鑒定到大量糖肽;且無非糖肽的非特異吸附。表明材料具有較好的糖肽富集能力。
實施例5
1、烷基化硅球的制備:在50mL的圓底燒瓶中,加入25mL甲苯,加入700mg粒徑為5μm的硅膠顆粒,0.3ml的3-巰丙基三乙氧基硅烷,超聲2分鐘分散均勻,在燒瓶上接冷凝管,機械攪拌,保持400rad/min速度。反應裝置放在油浴鍋中,加熱回流15h,停止反應,冷卻至室溫。高速離心機3500rad/min的速度離心,去除上清液,依次使用甲苯、丙酮、甲醇、丙酮抽濾洗滌,重復抽濾洗滌3遍洗三遍,50℃真空干燥箱內真空干燥24小時,得到修飾的硅烷化試劑的硅膠顆粒。
2、親水固定相的制備:在50mL的圓底燒瓶中,加入反應溶劑水-乙醇混合溶液(體積比為2:1)25mL,加入500mg修飾硅烷化的硅膠顆粒、150mg N-[三(羥甲基)甲基]丙烯酰胺(THMA)、100mg甲叉雙丙烯酰胺酸、7mg偶氮二異丁腈(AIBN)超聲1分鐘,使得加入的試劑以及顆粒溶解形成均勻分散在溶劑中,之后通氮氣5分鐘,機械攪拌,保持400rad/min速度。反應裝置放在油浴鍋中均勻加熱,在30min內升溫至75℃。維持75℃條件下反應24小時,停止反應,冷卻至室溫,得到微球顆粒silica@(MBAAm-co-THMA)之后使用高速離心機用4000rad/min的速度離心,去除上清液,加入反應溶液洗三遍,50℃真空干燥箱內真空干燥24小時。得到固定相顆粒silica@(MBAAm-co-THMA)。
- 還沒有人留言評論。精彩留言會獲得點贊!