一種負載型催化劑及其制備方法和應用以及甲烷干重整制合成氣的方法與流程
本發明涉及負載型催化劑的研究領域,具體地,涉及一種負載型催化劑、一種負載型催化劑的制備方法、由該方法制備得到的負載型催化劑、所述負載型催化劑在甲烷干重整反應中的應用以及甲烷干重整制合成氣的方法。
背景技術:
甲烷干重整反應以溫室氣體CH4和CO2為原料,制備具有較低H2/CO比的合成氣,非常適合作為費托合成制長鏈烴、氨合成、烷基化反應、甲醇合成等工業過程的原料。該過程不僅可以實現CO2的資源化利用,更為甲烷的高效利用提供了一條有效途徑。因此,若能實現該工藝的商業化應用,不僅對于緩解能源危機,改變某些化工產品的生產過程和原料路線具有重大的現實意義,而且對于減少溫室氣體的排放,減輕“溫室效應”造成的對全球生態環境的破壞具有深遠的歷史意義。Ni基催化劑在甲烷干重整反應中表現出可以和貴金屬相媲美的活性,但是催化劑存在著嚴重的因積碳和燒結而快速失活的問題,尤其是在高溫反應過程中,催化劑中活性金屬會不斷遷移聚集而長大,導致催化劑活性不斷降低并加劇積炭的發生。因此,如何能使催化劑中活性金屬保持穩定,防止其在高溫反應過程中發生遷移聚集并導致金屬顆粒尺寸長大是制備高活性、高穩定性Ni基催化劑的關鍵。
為了制備具有穩定結構的Ni基催化劑,人們通常采用共沉淀方法來制備催化劑(Journal of Catalysis,249(2007)300);Catalysis Today,45(1998)35),這樣可以使得活性金屬Ni在整個催化劑體相空間內呈均勻分布,并 利用其它組分作為空間阻隔劑以防止活性金屬在高溫反應過程遷移聚集。但共沉淀方法操作步驟冗長,過程變量較多,催化劑重復性難以保證。相比較而言,浸漬法是一種較為簡便的催化劑制備方法,也是工業上應用最為廣泛的催化劑制備方法。但傳統浸漬方法制備的催化劑活性金屬分散度低,金屬晶粒尺寸大,催化劑活性低、穩定性差。因此,開發一種高效且簡便易行的催化劑勢在必行。
技術實現要素:
本發明的目的在于克服現有技術中甲烷干重整催化劑的活性穩定性低和抗積炭性能差的缺陷,提供了一種具有高活性和穩定性以及良好的抗積炭性能的新的負載型催化劑及其制備方法和應用以及甲烷干重整制合成氣的方法。
具體地,本發明提供了一種負載型催化劑,其中,所述催化劑含有載體以及負載在載體上的活性金屬組分和助劑,其中,所述活性金屬組分為Ni組分和/或Co組分,所述活性金屬組分的分散度為6-15%。
本發明還提供了一種負載型催化劑的制備方法,該方法包括,在表面活性劑存在下,將浸漬溶液與載體接觸,然后進行干燥和焙燒,其中,所述浸漬溶液中含有活性金屬組分的可溶性化合物和助劑的可溶性化合物。
本發明還提供了由上述方法制得的負載型催化劑。
本發明還提供了所述負載型催化劑在甲烷干重整反應中的應用。
本發明還提供了一種甲烷干重整制合成氣的方法,該方法包括在甲烷干重整制合成氣條件下,將甲烷和二氧化碳與催化劑接觸,其中,所述催化劑為本發明制備的上述負載型催化劑。
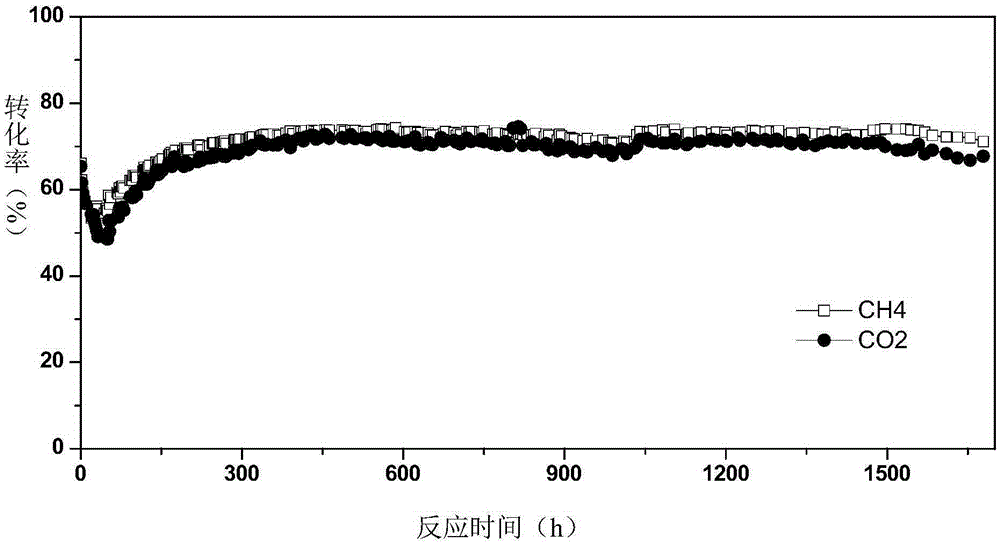
本發明提供的負載型催化劑和本發明提供的制備方法制得的負載型催化劑能夠得到顯著提高的活性金屬組分的分散度和較小的活性金屬晶粒尺 寸,從而大大提高了催化活性和穩定性以及抗積炭性能。本發明中的催化劑具有良好的性能的原因可能是:在催化劑制備過程中將活性金屬與助劑以共浸漬的方式同時負載到載體上,通過高溫處理使二者形成復合金屬氧化物新物相結構,從而使得所制備的催化劑中活性金屬分散度高、晶粒尺寸小,并且由于助劑的空間阻隔作用,可以有效防止活性金屬組分在高溫反應過程中的遷移聚集;從而保持其穩定性、高催化活性和抗積炭性能。從實施例1和對比例1的催化劑的反應性能對比圖可以看出,本發明提供的催化劑能在超高空速下(120000ml·g-1·h-1)高活性地連續穩定運行超過1500小時不失活。
本發明的其它特征和優點將在隨后的具體實施方式部分予以詳細說明。
附圖說明
附圖是用來提供對本發明的進一步理解,并且構成說明書的一部分,與下面的具體實施方式一起用于解釋本發明,但并不構成對本發明的限制。在附圖中:
圖1是實施例1所得的催化劑催化甲烷干重整反應的反應性能;
圖2是對比例1所得的催化劑催化甲烷干重整反應的反應性能;
圖3是對比例2所得的催化劑催化甲烷干重整反應的反應性能。
具體實施方式
以下對本發明的具體實施方式進行詳細說明。應當理解的是,此處所描述的具體實施方式僅用于說明和解釋本發明,并不用于限制本發明。
本發明提供了一種負載型催化劑,其中,所述催化劑含有載體以及負載在載體上的活性金屬組分和助劑,其中,所述活性金屬組分為Ni組分和/或 Co組分,所述活性金屬組分的分散度為6-15%。為了得到更好的催化活性和抗積炭性能,優選地,所述活性金屬組分的分散度為10-14%。
在本發明中,所述活性金屬組分的顆粒平均粒徑可以為2-50nm,優選為2-10nm。
根據本發明提供的負載型催化劑,所述活性金屬組分的含量可以參照現有技術進行確定。例如,以所述催化劑的總重量為基準,以金屬元素計,所述活性金屬組分的含量可以為2-20重量%,優選為3-15重量%,進一步優選為4-10重量%。需要說明的是,由于活性金屬組分實際以氧化物形式存在,而上述活性金屬組分以金屬元素的含量計,由此導致活性金屬組分的含量比實際小。顯然,當所述催化劑僅含有上述活性金屬組分、助劑和載體的時候,以氧化物計的活性金屬組分、助劑和載體的含量必然滿足100%。
本發明中,活性金屬組分的含量采用ICP法測得。
在本發明中,以金屬原子計,所述助劑與所述活性金屬組分的摩爾比可以為0.01-5:1,優選為0.1-2:1。
在本發明中,所述助劑的種類為本領域的常規選擇。例如,所述助劑可以為金屬氧化物助劑,優選為堿土和/或稀土金屬氧化物助劑;進一步優選為MgO、CaO、BaO、La2O3、CeO2、Sm2O3、ZrO2和Y2O3中的至少一種。
根據本發明,所述載體的種類沒有特別的限定,可以為本領域的常規選擇。例如,所述載體可以為單組分氧化物載體或者雙組分或三組分復合氧化物載體。優選情況下,所述載體選自SiO2、TiO2、MgO、Al2O3、ZrO2、CeO2、La2O3、SiO2-Al2O3、TiO2-SiO2、Al2O3-MgO和TiO2-SiO2-Al2O3中的一種或多種。
在本發明中,所述載體的形狀為本領域的常規選擇。例如,所述載體的形狀可以是圓柱形、球形、三葉草、四葉草、碟形和拉西環中的至少一種, 優選為四葉草和/或拉西環形狀。
本發明還提供了一種負載型催化劑的制備方法,該方法包括,在表面活性劑存在下,將浸漬溶液與載體接觸,然后進行干燥和焙燒,其中,所述浸漬溶液中含有活性金屬組分的可溶性化合物和助劑的可溶性化合物。
在本發明中,所述表面活性劑的用量沒有特別的限定。但是為了形成活性更高穩定性更好的催化劑,所述表面活性劑和以金屬原子計的活性金屬組分的可溶性化合物的用量的摩爾比可以為0.001-2:1,優選為0.001-1:1,進一步優選為0.01-0.8:1。
在本發明中,所述表面活性劑的種類可以為本領域的常規選擇。例如,所述表面活性劑可以選自陰離子型表面活性劑、兩性表面活性劑和非離子型表面活性劑中的至少一種;優選為硬脂酸、油酸、月桂酸、卵磷脂、十二烷基氨基丙酸、烷基二甲基甜菜堿、脂肪酸甘油酯、多元醇、吐溫60和P123中的至少一種;進一步優選為P123、油酸和吐溫60中的至少一種。
在本發明中,所述活性金屬組分的可溶性化合物的用量沒有特別的限定,可以為本領域的常規選擇。例如,為了使所得催化劑中,以所述催化劑的總重量為基準,以金屬元素計,所述活性金屬組分的含量為2-20重量%,優選為3-15重量%,進一步優選為4-10重量%,相對于100重量份的載體,所述活性金屬組分的可溶性化合物的用量可以為10-100重量份,優選為15-75重量份,更優選為20-50重量份。
根據本發明,所述活性金屬組分的可溶性化合物的種類為本領域技術人員熟知,可以為本領域的常規選擇。例如,所述活性金屬組分的可溶性化合物可以選自Ni(NO3)2·6H2O、Co(NO3)2·6H2O、Ni(acac)2、Co(acac)3、Ni(CH3COO)2·6H2O和Co(CH3COO)2·6H2O中的至少一種,優選為Ni(NO3)2·6H2O和/或Co(NO3)2·6H2O。
根據本發明,所述載體可以為單組分氧化物載體或者雙組分或三組分復合氧化物載體。優選情況下,所述載體選自SiO2、TiO2、MgO、Al2O3、ZrO2、CeO2、La2O3、SiO2-Al2O3、TiO2-SiO2、Al2O3-MgO和TiO2-SiO2-Al2O3中的一種或多種。
根據本發明,在所述浸漬溶液中,以金屬元素計,活性金屬組分的可溶性化合物和助劑的可溶性化合物的總濃度可以為21.8-162.8克/升,載體的用量使得所得催化劑中以催化劑的總重量為基準,以金屬元素計的所述活性金屬組分的含量為2-20重量%,優選為3-15重量%,進一步優選為4-10重量%。
在本發明中,所述助劑的用量沒有特別的限定。例如,以金屬原子計,所述助劑的可溶性化合物與所述活性金屬組分的可溶性化合物的摩爾比可以為0.01-5:1,優選為0.1-2:1。
在本發明中,所述助劑的可溶性化合物的種類為本領域的常規選擇。例如,所述助劑的可溶性化合物可以選自堿土和/或稀土金屬可溶性化合物;優選地,所述助劑的可溶性化合物選自鎂鹽、鈣鹽、鍶鹽、鋇鹽、鈰鹽、鑭鹽、鋯鹽和釔鹽中的至少一種;更優選地,所述助劑的可溶性化合物選自鎂鹽、鋇鹽、鑭鹽和釔鹽中的至少一種,進一步優選為鑭鹽和/或鎂鹽。
根據本發明,所述浸漬溶液與載體接觸的條件沒有特別的限定。例如,所述浸漬溶液與載體接觸的條件包括:溫度可以為10-50℃,優選為15-30℃;時間可以為0.5-10小時,優選為2-5小時。
本發明對所述浸漬溶液與載體的接觸方式沒有特別的限定。例如,可以先將表面活性劑與含有活性金屬組分的可溶性化合物的浸漬溶液混合,然后與載體接觸。
在本發明中,對浸漬溶液與載體的接觸后的產物進行干燥和焙燒,其中,干燥和焙燒的條件為本領域技術人員熟知。例如,干燥的條件包括:溫度可 以為80-140℃,優選為100-120℃;時間可以為1-10小時,優選為5-10小時。所述焙燒的溫度可以為400-1000℃,優選為500-800℃;時間可以為1-10小時,優選為2-6小時。
本發明還提供了由上述方法制備得到的負載型催化劑。
本發明還提供了所述負載型催化劑在甲烷干重整制備合成氣中的應用。
根據本發明提供的方法制備的催化劑用于甲烷干重整反應時,反應之前需要在氫氣存在下,將活性金屬進行還原活化。其中,還原活化的條件包括:還原溫度可以為300-800℃,優選為400-750℃,還原時間可以為0.5-10小時,優選為1-5小時;所述還原活化可以在純氫中進行,也可在氫氣和惰性氣體的混合氣中進行,如果在氫氣與氮氣和/或氬氣的混合氣中進行,還原壓力可以為0-2MPa,優選為0-1MPa。本發明中,所述壓力為表壓。
本發明還提供了一種甲烷干重整制合成氣的方法,該方法包括在甲烷干重整制合成氣條件下,將甲烷和二氧化碳與催化劑接觸,其中,所述催化劑為本發明上述負載型催化劑。
其中,甲烷和二氧化碳與所述催化劑接觸的方法沒有特別的限定,可以為本領域的常規選擇,例如,可以將甲烷和二氧化碳各自送入反應器中同時與催化劑接觸,也可以將甲烷和二氧化碳形成混合物再與所述催化劑接觸,優選地,將甲烷和二氧化碳形成混合物再與所述催化劑接觸。
按照本發明提供的載體制備的催化劑用于催化甲烷和CO2反應制備合成氣時,采用固定床反應器或流化床反應器。所述甲烷干重整制合成氣的條件包括:甲烷和二氧化碳的摩爾比可以為0.7-1.1:1,優選為0.8-1:1;反應溫度可以為550-850℃,優選為600-800℃;壓力可以為0-3MPa,優選為0-1MPa;原料氣的總空速可以為2000-120000ml·g-1·h-1,優選為60000-120000ml·g-1·h-1。
以下將通過實施例對本發明進行詳細描述。
以下實施例中,產品的性能測試采用以下方法進行:
1)金屬分散度由氫氣化學吸附法采用Micromeritics(ASAP-2010C)化學吸附儀進行測量。具體的,將0.2g樣品先經300℃脫氣處理1小時,然后升溫至700℃還原2小時,再降溫至40℃進行氫氣化學吸附操作。之后根據化學吸附氫氣的量通過下述公式計算活性金屬組分的分散度和金屬顆粒的平均粒徑;
活性金屬分散度D:
活性金屬顆粒的平均粒徑d:
其中Vad是指標準狀態下氫氣的單層吸附量,單位為mL;Ws是樣品質量,單位為g;FWMe是金屬Me的摩爾質量,單位為g/mol;FMe是催化劑中金屬的負載量,單位為%;Vm是指標態下的摩爾氣體體積,單位為mL/mol;SAMe是金屬的比表面積,單位為m2/g;ρMe是金屬的密度,單位為kg/m3;
2)利用氣相色譜法在線取樣分析計算尾氣組成;
3)活性金屬組分的含量采用ICP法測得。
實施例1
該實施例用于說明本發明提供的催化劑和催化劑的制備方法及其應用。
(1)催化劑制備
將1.765g的Ni(NO3)2·6H2O和1.167g的Mg(NO3)2·6H2O溶于8.4ml去離子水中攪拌溶解,然后加入0.61g的P123,混合均勻得到浸漬溶液。取 4g的SiO2載體分散到浸漬溶液中,溫度為25℃下靜置2小時后,蒸干水分,然后置于烘箱中120℃干燥10小時。干燥后的樣品置于馬弗爐中600℃焙燒3小時,所得催化劑記為6Mg-Ni/SiO2。以金屬原子計,所述助劑與所述活性金屬組分的摩爾比為0.75:1,以金屬元素計的活性金屬組分Ni的含量為8重量%。該催化劑中所述活性金屬組分的分散度為12.7%,活性金屬組分的顆粒平均粒徑為5.4nm。
(2)催化劑評價
稱取上述6Mg-Ni/SiO2催化劑0.1g,用40-60目石英砂稀釋至2ml,裝入內徑8mm的石英管反應器中,常壓下于純氫氣氛中700℃還原3小時進行活化。還原結束后,在氫氣氣氛下升溫至750℃,切換原料氣(CH4/CO2=1/1)進行反應,反應空速為120000ml·g-1·h-1,反應壓力為常壓。反應穩定進行80小時后,由氣相色譜在線取樣并分析尾氣組成。計算得到:XCH4=62.9%,XCO2=61.5%,H2/CO=0.96。
實施例1中所得的催化劑的穩定性評價結果列于圖1中,具體為反應時間為0-1700個小時的所述催化劑所催化的甲烷干重整反應中的甲烷和二氧化碳轉化率。
實施例2
該實施例用于說明本發明提供的催化劑和催化劑的制備方法及其應用。
(1)催化劑制備
將0.873g的Ni(NO3)2·6H2O、0.872g的Co(NO3)2·6H2O和1.955g的La(NO3)3·6H2O溶于8.4ml去離子水中攪拌溶解,然后加入1.57g的吐溫60,混合均勻得到浸漬溶液。取4g的SiO2載體分散到浸漬溶液中,溫度為30℃下靜置2小時后,蒸干水分,然后置于烘箱中100℃干燥7小時。干燥后的 樣品置于馬弗爐中800℃焙燒2小時,所得催化劑記為4La-Ni-Co/SiO2。以金屬原子計,所述助劑與所述活性金屬組分的摩爾比為0.75:1,以金屬元素計的所述活性金屬Ni和Co的總含量為8重量%。該催化劑中所述活性金屬Ni組分的分散度為10.9%,活性金屬組分的顆粒平均粒徑為9.1nm。
(2)催化劑評價
在與實施例1相同的條件下活化催化劑并進行甲烷干重整反應。反應穩定進行80小時后,由氣相色譜在線取樣并分析尾氣組成。計算得到:XCH4=65.4%,XCO2=66.7%,H2/CO=1.02。
實施例3
該實施例用于說明本發明提供的催化劑和催化劑的制備方法及其應用。
(1)催化劑制備
將2.81g的Ni(NO3)2·6H2O和0.31g的Mg(NO3)2·6H2O溶于8.4ml去離子水中攪拌溶解,然后加入1.36g的油酸,混合均勻得到浸漬溶液。取4g的SiO2載體分散到浸漬溶液中,溫度為15℃下靜置5小時后,蒸干水分,然后置于烘箱中110℃干燥5小時。干燥后的樣品置于馬弗爐中500℃焙燒6小時,所得催化劑記為1Mg-Ni/SiO2。以金屬原子計,所述助劑與所述活性金屬組分的摩爾比為0.125:1,以金屬元素計的所述活性金屬Ni的含量為12重量%。該催化劑中所述活性金屬組分的分散度為12.4%,活性金屬組分的顆粒平均粒徑為6.1nm。
(2)催化劑評價
在與實施例1相同的條件下活化催化劑并進行甲烷干重整反應。反應穩定進行80小時后,由氣相色譜在線取樣并分析尾氣組成。計算得到:XCH4=58.6%,XCO2=59.7%,H2/CO=1.01。
實施例4
該實施例用于說明本發明提供的催化劑和催化劑的制備方法及其應用。
(1)催化劑制備
按照實施例1中的方法制備催化劑,所不同的是,P123的用量為0.348g,所得催化劑記為6Mg-Ni/SiO2-2。該催化劑中所述活性金屬組分的分散度為11.6%,活性金屬組分的顆粒平均粒徑為5.8nm。
(2)催化劑評價
在與實施例1相同的條件下活化催化劑并進行甲烷干重整反應。反應穩定進行80小時后,由氣相色譜在線取樣并分析尾氣組成。計算得到:XCH4=59.3%,XCO2=60.4%,H2/CO=1.02。
實施例5
該實施例用于說明本發明提供的催化劑和催化劑的制備方法及其應用。
(1)催化劑制備
按照實施例1中的方法制備催化劑,所不同的是,Mg(NO3)2·6H2O的用量為3.08g,所得催化劑記為10Mg-Ni/SiO2-3。以金屬原子計,所述助劑與所述活性金屬組分的摩爾比為2:1,以金屬元素計的活性金屬組分Ni的含量為7.4重量%。該催化劑中所述活性金屬組分的分散度為13.5%,活性金屬組分的顆粒平均粒徑為4.7nm。
(2)催化劑評價
在與實施例1相同的條件下活化催化劑并進行甲烷干重整反應。反應穩定進行80小時后,由氣相色譜在線取樣并分析尾氣組成。計算得到:XCH4=64.6%,XCO2=62.9%,H2/CO=1.01。
實施例6
該實施例用于說明本發明提供的催化劑和催化劑的制備方法及其應用。
(1)催化劑制備
按照實施例1中的方法制備催化劑,所不同的是,載體選用氧化鎂,所得催化劑記為6Mg-Ni/MgO。該催化劑中所述活性金屬組分的分散度為13.1%,活性金屬組分的顆粒平均粒徑為4.9nm。
(2)催化劑評價
在與實施例1相同的條件下活化催化劑并進行甲烷干重整反應。反應穩定進行80小時后,由氣相色譜在線取樣并分析尾氣組成。計算得到:XCH4=63.7%,XCO2=61.9%,H2/CO=1.0。
實施例7
該實施例用于說明本發明提供的催化劑和催化劑的制備方法及其應用。
(1)催化劑制備
按照實施例1中的方法制備催化劑,所不同的是,載體選用Al2O3-MgO復合載體,所得催化劑記為6Mg-Ni/Al2O3-MgO。該催化劑中所述活性金屬組分的分散度為13.9%,活性金屬組分的顆粒平均粒徑為4.2nm。
(2)催化劑評價
在與實施例1相同的條件下活化催化劑并進行甲烷干重整反應。反應穩定進行80小時后,由氣相色譜在線取樣并分析尾氣組成。計算得到:XCH4=66.7%,XCO2=65.1%,H2/CO=1.01。
對比例1
該對比例用于說明參比的催化劑和催化劑的制備方法及其應用。
(1)催化劑制備
按照實施例1中的方法制備催化劑,所不同的是,不使用表面活性劑 P123,所得催化劑記為6Mg-Ni/SiO2-D1。該催化劑中所述活性金屬組分的分散度為2.3%,活性金屬組分的顆粒平均粒徑為44.1nm。
(2)催化劑評價
在與實施例1相同的條件下活化催化劑并進行甲烷干重整反應。反應穩定進行28小時后,由氣相色譜在線取樣并分析尾氣組成。計算得到:XCH4=16.3%,XCO2=21.1%,H2/CO=0.98。
對比例1中所得的催化劑的反應性能列于圖2,具體為反應時間為0-30小時的所述催化劑所催化的甲烷干重整反應中的甲烷和二氧化碳轉化率。
對比例2
該對比例用于說明參比的催化劑和催化劑的制備方法及其應用。
(1)催化劑制備
按照實施例1中的方法制備催化劑,所不同的是,不使用助劑,所得催化劑記為Ni/SiO2。該催化劑中所述活性金屬組分的分散度為5.4%,活性金屬組分的顆粒平均粒徑為18.7nm。
(2)催化劑評價
在與實施例1相同的條件下活化催化劑并進行甲烷干重整反應。反應穩定進行80小時后,由氣相色譜在線取樣并分析尾氣組成。計算得到:XCH4=32.2%,XCO2=21.0%,H2/CO=1.02。
對比例2中所得的催化劑的反應性能列于圖3,具體為反應時間為0-100小時的所述催化劑所催化的甲烷干重整反應中的甲烷和二氧化碳轉化率。
從上述結果可以看出,使用本發明提供的催化劑的制備方法以及制備得到的催化劑能連續高效穩定運行超過1500個小時,而且在120000ml·g-1·h-1的高空速下還能獲得較高的甲烷和二氧化碳轉化率。由此說明本發明的催化 劑具有更好的反應活性和穩定性以及抗積炭性能。
從實施例1-5的結果可以看出,即便是反應活性較低的二氧化硅載體,使用本發明的方法制成催化劑后,也能夠獲得活性金屬組分的分散度為6-15%,催化劑具有更好的反應活性和穩定性以及抗積炭性能,能連續高效穩定運行超過1500個小時。
從實施例1和對比例1和對比例2的結果可以看出,使用本發明提供的催化劑的制備方法以及制備得到的催化劑具有更好的反應活性和穩定性以及抗積炭性能,能連續高效穩定運行超過1500個小時。而未采用本發明方法的對比例1和對比例2的催化劑反應活性低,穩定性差,運行30個小時活性就大幅下降。
以上詳細描述了本發明的優選實施方式,但是,本發明并不限于上述實施方式中的具體細節,在本發明的技術構思范圍內,可以對本發明的技術方案進行多種簡單變型,這些簡單變型均屬于本發明的保護范圍。
另外需要說明的是,在上述具體實施方式中所描述的各個具體技術特征,在不矛盾的情況下,可以通過任何合適的方式進行組合。為了避免不必要的重復,本發明對各種可能的組合方式不再另行說明。
此外,本發明的各種不同的實施方式之間也可以進行任意組合,只要其不違背本發明的思想,其同樣應當視為本發明所公開的內容。
- 還沒有人留言評論。精彩留言會獲得點贊!