石墨烯-貴金屬納米顆粒復合水、氣凝膠及其制備方法、應用與流程
本發明涉及一種石墨烯-貴金屬無機納米顆粒復合水凝膠、氣凝膠及其制備方法、應用。
背景技術:
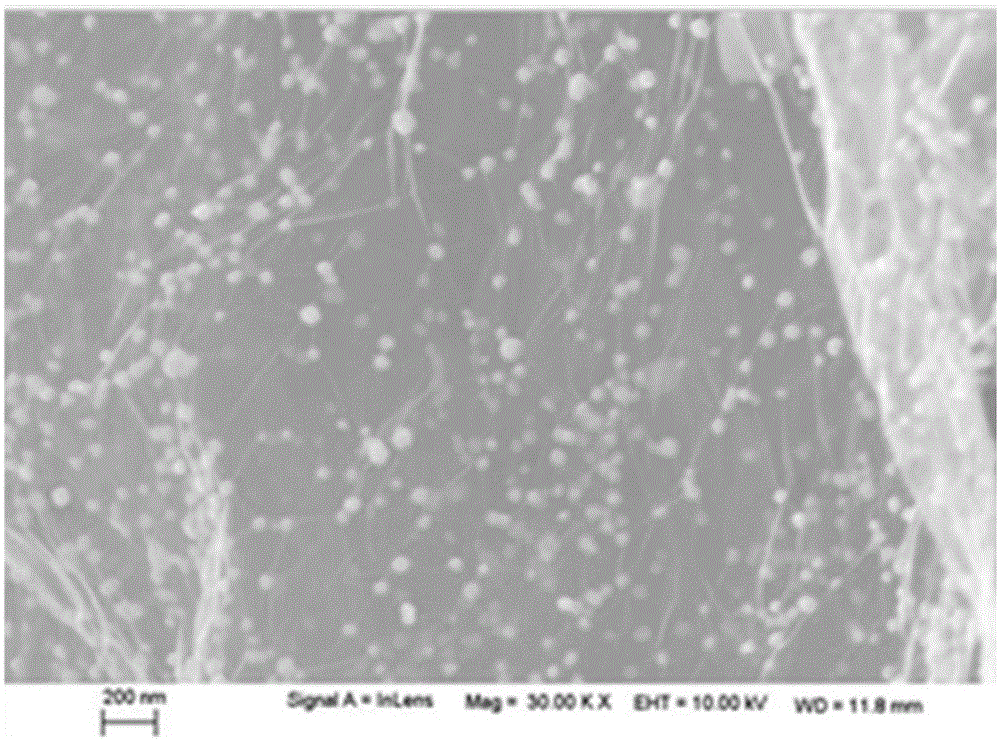
:從實際應用角度考慮,將納米石墨烯材料轉變為宏觀結構材料無疑是非常有價值的研究方向。而將多功能性的貴金屬無機納米顆粒與宏觀石墨烯結構復合制備宏觀石墨烯-貴金屬無機納米顆粒復合材料無疑將會給工業界帶來較大的有利價值。貴金屬包括Au、Ag和鉑族金屬Ru、Rh、Pd、Os、Ir以及Pt等幾種價格昂貴的金屬。貴金屬納米粒子具有區別于本體材料的優異的光、電、磁和催化性能,具有極大的比表面積,相當強的導電性和反應性,表面活性位點多等特點,是一類優良的電分析和電催化的納米材料,在化學和生物等眾多領域得到廣泛的應用。目前,針對宏觀石墨烯結構材料(尤其是宏觀三維石墨烯基塊體凝膠材料)或貴金屬與石墨烯的粉體材料的研究和制備已經有大量的研究,而宏觀塊體石墨烯-貴金屬無機納米顆粒復合氣凝膠材料的制備和研究鮮有報道。例如中國專利CN104250005A公開了一種石墨烯氣凝膠及其制備方法和應用,該專利中制備的單純的石墨烯氣凝膠的應用范圍是有限的,沒有將多功能的貴金屬納米顆粒與宏觀的石墨烯氣凝膠相結合,在實際應用中受到一定的限制;高超等報道了(ScienceChinaChemistry,2011,54(2):397-404.)一種貴金屬(如Ag、Au、Pt和Pd)和石墨烯的粉體材料,該材料以石墨烯為載體與貴金屬納米顆粒復合用于催化反應,但是其仍然屬于納米粉體材料,而納米粉體材料的共同缺點就是容易團聚,在實際應用中存在應用效率低,回收困難的缺陷。若將石墨烯與貴金屬納米顆粒進行直接共混進行復合材料的制備,該制 備方法會使得貴金屬納米顆粒團聚嚴重,致使貴金屬無機納米顆粒自身良好的催化、電學性能等優異特性,因團聚而使其利用效率受限,無法為高速發展的應用產業提供選擇。如何提高貴金屬納米顆粒在石墨烯復合材料中的高度分散直接影響石墨烯-貴金屬納米顆粒復合材料的應用潛能。因此,針對上述問題,一種一步法原位制備貴金屬納米顆粒在石墨烯氣凝膠材料中分散性好的、緊密結合但不團聚的石墨烯-貴金屬無機納米顆粒復合凝膠材料的制備方法有待開發,其對石墨烯的應用研究及市場化生產具有十分重要的意義。技術實現要素:本發明克服了現有技術中石墨烯-貴金屬無機納米顆粒復合氣凝膠的制備和研究鮮有報道,無法為高速發展的應用產業提供選擇的缺陷,提供了一種石墨烯-貴金屬無機納米顆粒復合水凝膠、氣凝膠及其制備方法、應用。本發明采用氧化石墨烯與貴金屬水溶性化合物水溶液為前驅體,借助氧化石墨烯的引入,使得水溶性貴金屬粒子十分均勻地分散并吸附在氧化石墨烯表面,避免了納米粒子的聚集,利用輻照技術原位還原自組裝制備了石墨烯-貴金屬納米顆粒復合氣凝膠,操作簡單,綠色環保,目前尚無報道;且該復合氣凝膠為多孔、大孔結構,孔徑分布在10-100μm之間,結構較均勻,貴金屬納米顆粒均勻分布在石墨烯氣凝膠框架上,結合了石墨烯氣凝膠與貴金屬納米顆粒的功能,在吸附有機溶劑的過程中,具有良好的電導性,可以為相應的電催化反應提供可行性,使其在小分子電分析和電催化等有機反應催化合成領域、石油有機中間體的合成領域等方面均具有巨大的潛在應用價值。本發明通過以下技術方案解決上述技術問題。本發明提供了一種石墨烯-貴金屬無機納米顆粒復合水凝膠的制備方法,其包括下述步驟:(1)將氧化石墨烯分散液、水溶性貴金屬化合物以及水溶性還原劑混合均勻得氧化石墨烯混合液;其中,所述的氧化石墨烯混合液中,氧化石墨烯與所述的水溶性貴金屬化合物的質量比為(1:0.01)-(1:5);所述水溶性還原劑包括醇類水溶性還原劑和/或胺類水溶性還原劑;當所述水溶性還原劑含有所述醇類水溶性還原劑時,所述醇類水溶性還原劑占所述氧化石墨烯混合液的質量百分比為2-90%;當所述水溶性還原劑含有所述胺類水溶性還原劑時,在所述的氧化石墨烯混合液中,氧化石墨烯與所述胺類水溶性還原劑的質量比為(1:0.5)-(1:200);(2)將所述氧化石墨烯混合液用高能射線照射進行輻照反應得石墨烯-貴金屬無機納米顆粒復合水凝膠。其中,步驟(1)中,所述的氧化石墨烯分散液由本領域內常規方法制得,較佳地由氧化剝離石墨法(即Hummers法)制得,更佳地通過下述步驟制得:①預氧化:將9-11g石墨、130-160mL濃硫酸和40-60mL濃硝酸倒入水中,室溫攪拌22-26h,加入0.8-1.2L去離子水中,過濾,烘干;重復上述預氧化過程2-3次,得到預氧化石墨;②熱膨脹:將步驟①的預氧化石墨在400-900℃條件下熱膨脹10-30s,得到熱膨脹氧化石墨;③將步驟②的熱膨脹氧化4-6g石墨與280-320mL濃硫酸、4.1-4.3gK2S2O8和6.1-6.3g五氧化二磷的混合物在80-90℃條件下加熱,加入水過濾洗滌,干燥,得到預氧化熱膨脹石墨;④將步驟③的預氧化熱膨脹石墨4-6g與180-220mL濃硫酸在0-5℃條件下混合,加入14-16g高錳酸鉀,34-36℃反應1.5-2.5h,再加入18-22mL雙氧水,靜置,離心洗滌,加入水攪拌,即可。其中,步驟(1)中,所述的水溶性貴金屬化合物為本領域內常規,較佳地為水溶性貴金屬酸和/或水溶性貴金屬鹽。所述水溶性貴金屬酸為本領域內常規,較佳地為氯金酸、氯鉑酸、氯鈀酸和氯銠酸中的一種或多種。所述水溶性貴金屬鹽為本領域內常規,較佳地為硝酸銀、氯釕酸鉀、氯銠酸鉀、三氯化釕、三氯化銠和三氯化銥中的一種或多種。所述水溶性是指能夠在不同溫度的水中溶解即可。其中,步驟(1)中,水溶性貴金屬化合物添加量過低,制備的復合氣凝膠體現不出貴金屬優異的性能;添加量過高,則宏觀復合氣凝膠材料較難成型。故在所述的氧化石墨烯混合液中,氧化石墨烯與所述的水溶性貴金屬化合物的質量比較佳地為(1:0.05)-(1:3)。其中,步驟(1)中,所述的醇類水溶性還原劑為本領域內常規,較佳地為甲醇、乙醇、丙醇、異丙醇、丁醇、丁二醇、異丁醇、乙二醇、丙二醇、丙三醇、辛二醇和聚乙烯醇中的一種或多種。其中,步驟(1)中,所述的胺類水溶性還原劑為本領域內常規,較佳地為甲胺、乙二胺、丙二胺、丁二胺、二乙烯三胺、三乙烯四胺、四乙烯五胺、聚烯丙基胺、N,N'-二(2-氨乙基)-1,3-丙二胺和氨中的一種或多種。其中,步驟(1)中,所述氧化石墨烯混合液中,氧化石墨烯的含量較佳地為1-20mg/mL,更佳地為2-10mg/mL。其中,步驟(1)中,若水溶性還原劑的含量太少,得到的是懸浮在管內的石墨烯-貴金屬無機納米復合顆粒,得不到具有一定自支撐力學強度的連續濕態石墨烯-貴金屬無機納米復合水凝膠;若水溶性還原劑的含量較多,不僅增加成本還致使氧化石墨烯以及貴金屬離子在溶液中的分散性較差,不容易得到連續的石墨烯-貴金屬無機納米顆粒復合水凝膠。由此,步驟(1)中,當所述水溶性還原劑含有所述醇類水溶性還原劑時,為了實現石墨烯-貴金屬無機納米顆粒復合氣凝膠的成型效果,所述醇類水溶性還原劑占所述氧化石墨烯混合液的質量百分比較佳地為5-90%,更佳地為10-80%。而當所述水溶性還原劑含有所述胺類水溶性還原劑時,為了更好地實現石墨烯-貴金屬無機納米顆粒復合氣凝膠的成型效果,在所述的氧化石墨烯混合液中,氧化石墨烯與所述胺類水溶性還原劑的質量比較佳地為(1:1)-(1:150),更佳地為(1:5)-(1:100)。其中,步驟(2)中,所述輻照反應的氣氛一般為無氧氣氛或空氣氣氛,為了更佳地實現石墨烯-貴金屬無機納米顆粒復合氣凝膠的成型效果,較佳 地為無氧氣氛。所述的無氧氣氛較佳地為氮氣和/或氬氣。其中,步驟(2)中,所述的高能射線較佳地為γ射線或電子束射線。其中,步驟(2)中,所述的輻照反應的劑量較佳地為50-800kGy,更佳地為110-600kGy。其中,步驟(2)中,為了實現石墨烯-貴金屬無機納米顆粒復合氣凝膠的成型效果,所述的輻照反應的劑量率較佳地為0.1-15kGy/小時,更佳地為1-15kGy/小時。本發明還提供了一種由上述制備方法制得的石墨烯-貴金屬無機納米顆粒復合水凝膠。其中,所述的石墨烯-貴金屬無機納米顆粒復合水凝膠中,貴金屬無機納米顆粒為本領域內常規,較佳地為金、鉑、鈀、銠、銀、釕和銥中的一種或多種。本發明還提供了一種石墨烯-貴金屬無機納米顆粒復合氣凝膠的制備方法,其包括下述步驟:將上述石墨烯-貴金屬無機納米顆粒復合水凝膠進行冷凍干燥或超臨界二氧化碳干燥,即可。其中,所述的冷凍干燥為本領域常規操作。其中,所述的超臨界二氧化碳干燥為本領域常規操作。本發明還提供了一種由上述制備方法制得的石墨烯-貴金屬無機納米顆粒復合氣凝膠。其中,所述的石墨烯-貴金屬無機納米顆粒復合氣凝膠為多孔、大孔結構,結構較均勻,孔徑分布在10-100μm之間。所述的石墨烯-貴金屬無機納米顆粒復合氣凝膠中,貴金屬無機納米顆粒的直徑較佳地為40-80nm。其中,所述的石墨烯-貴金屬無機納米顆粒復合氣凝膠中,貴金屬無機納米顆粒為本領域內常規,較佳地為金、鉑、鈀、銠、銀、釕和銥中的一種或多種。若貴金屬納米顆粒與石墨烯直接共混成型制備的復合氣凝膠中,貴金屬無機納米顆粒僅填充在石墨烯氣凝膠多孔結構的內部,在石墨烯表面是無法 負載的,而且其團聚嚴重,分散性差,致使其自身良好的催化、電學性能等優異特性,因團聚致使其利用效率受限。而本發明制得的石墨烯-貴金屬無機納米顆粒復合氣凝膠,與其截然不同。本發明的納米顆粒是由離子原位形成在石墨烯的表面,其均勻負載在石墨烯復合氣凝膠的框架結構上,包括外表面和內部多孔結構上,所以本發明的貴金屬納米粒子不僅僅分散性較好,而且其粘合在石墨烯片層上慢慢成型,兩者緊密結合而無團聚現象,使制備的復合氣凝膠在吸收有機溶劑的過程中,有利于充分發揮貴金屬無機納米粒子自身良好的催化、電學性能等有益特性,具有極大的潛在應用價值。本發明還提供了一種所述的石墨烯-貴金屬無機納米顆粒復合氣凝膠在吸附有機溶劑中的應用。在符合本領域常識的基礎上,上述各優選條件,可任意組合,即得本發明各較佳實例。本發明所用試劑和原料均市售可得。本發明的積極進步效果在于:1、本發明通過一步法輻照還原組裝并借助冷凍干燥方法直接得到石墨烯-貴金屬納米顆粒復合材料,輻照還原反應在環境溫度下進行,操作簡潔,有望實現技術產業化。2、本發明的原材料為氧化石墨烯和水溶性貴金屬化合物,來源廣泛,利用率較高,所用氧化石墨烯原料反應后完全凝膠成型,凝膠周圍水溶液為透明,原料幾乎無損失;選用水溶性醇和/或胺還原劑作為輻照反應體系,配制過程簡單,不涉及苛刻的化學反應條件,避免了復雜而困難的化學反應和純化過程。3、本發明在利用醇類水溶性還原劑做添加劑時得到的石墨烯-貴金屬無機納米顆粒復合氣凝膠較為純凈,除碳、氧、貴金屬元素外無其它元素摻雜;本發明在利用含胺類水溶性還原劑做添加劑時得到的石墨烯-貴金屬無機納米顆粒復合氣凝膠較為純凈,除碳、氮、氧、貴金屬元素外無其它元素摻雜。4、本發明的石墨烯-貴金屬無機納米顆粒復合氣凝膠的形狀和大小可通 過采用不同形狀和大小的輻照反應器進行調整;其密度可通過改變反應物濃度進行調控;其為多孔、大孔結構,孔徑分布在10-100μm之間,結構較為均勻;貴金屬納米顆粒分布非常均勻,基本無團聚現象;其比表面積大、導電性好、熱傳導性能優異。5、本發明的制備方法可以得到不同貴金屬種類的單一金屬納米顆粒-石墨烯復合氣凝膠,也可以得到兩種或兩種以上貴金屬復合的石墨烯-貴金屬納米顆粒復合氣凝膠材料。石墨烯氣凝膠的石墨烯片層提供了貴金屬納米顆粒的負載位點,為貴金屬納米顆粒的原位形成、均勻分散性和應用性提供了載體;石墨烯-貴金屬納米顆粒復合氣凝膠結合了石墨烯氣凝膠與貴金屬納米顆粒的功能,負載有貴金屬納米顆粒的石墨烯復合凝膠在吸附有機溶劑的過程中,具有良好的電導性,可以為相應的電催化反應提供可行性,使其在小分子電分析和電催化等有機反應催化合成領域、石油有機中間體的合成領域等方面均具有巨大的潛在應用價值。附圖說明圖1為實施例1制備的石墨烯-銀無機納米顆粒復合氣凝膠中多孔結構掃描電鏡(低倍)圖。圖2為實施例1制備的石墨烯-銀無機納米顆粒復合氣凝膠中銀納米顆粒在石墨烯片層上的負載掃描電鏡(高倍)圖。具體實施方式下面通過實施例的方式進一步說明本發明,但并不因此將本發明限制在所述的實施例范圍之中。下列實施例中未注明具體條件的實驗方法,按照常規方法和條件,或按照商品說明書選擇。實施例1下述實施例中,所用石墨由西格瑪奧德里奇(Sigma-Aldrich)公司提供, 所用石墨為鱗片石墨,平均粒徑為50-500μm,其余原料均由國藥集團化學試劑有限公司提供。實施例1(1)氧化石墨烯分散液的制備方法:石墨10g,98%濃硫酸150ml,65%的濃硝酸50ml,加入到500ml錐形瓶中室溫攪拌24h,慢慢倒入1L水中過濾收集固體,洗滌3次,80℃烘干4小時。重復預氧化過程兩次。將干燥后的預氧化石墨放入箱式爐中900℃熱膨脹10s得到熱膨脹氧化石墨。在500ml廣口錐形瓶中將5g熱膨脹氧化石墨與300ml98%的濃硫酸,4.2gK2S2O8,6.2g五氧化二磷混合后80℃加熱4小時,用2L水稀釋,過濾洗滌,空氣中干燥3天得到預氧化熱膨脹石墨。將干燥的預氧化熱膨脹石墨5g與200ml98%的濃硫酸在低溫0-5℃下混合,加入15g高錳酸鉀,慢慢加入,35℃攪拌2h,加2L水稀釋靜置1h后加入20ml30%的雙氧水,靜置2天,離心洗滌,加入水攪拌,即可,得到分散較好的氧化石墨烯分散液。(2)將步驟(1)制得的氧化石墨烯分散液、硝酸銀和丙二醇混合制備得1mg/ml氧化石墨烯混合溶液;其中,氧化石墨烯與硝酸銀的質量比為1:0.1,丙二醇占氧化石墨烯混合溶液的質量百分比為2%。(3)將步驟(2)得到的氧化石墨烯混合溶液注入圓柱狀輻照反應器中,通氮氣除氧;將封好的輻照反應器用鈷60γ射線源輻照得石墨烯-銀納米顆粒復合水凝膠;其中,劑量為300kGy,輻照反應的劑量率為3kGy/小時。(4)將步驟(3)所得石墨烯-銀納米顆粒復合水凝膠進行冷凍干燥,得干態的圓柱狀石墨烯-銀納米顆粒氣凝膠。圖1為制備的石墨烯-銀無機納米顆粒復合氣凝膠中多孔結構掃描電鏡(低倍)圖,發現其為多孔、大孔結構,結構較均勻,孔徑分布在10-100μm之間。圖2為制備的石墨烯-銀無機納米顆粒復合氣凝膠中銀納米顆粒在石墨烯片層上的負載掃描電鏡(高倍)圖,發現銀納米顆粒大小分布均勻,直徑在40-80nm。該石墨烯-銀無機納米顆粒復合氣凝膠具有良好的吸油性能, 在小分子電分析和電催化等有機反應催化合成領域、石油有機中間體的合成領域等方面均具有巨大的潛在應用價值。其碳氧比、密度和吸附容量如表1所示。實施例2(1)氧化石墨烯分散液的制備方法與實施例1相同。(2)將步驟(1)制得的氧化石墨烯分散液、氯金酸、三氯化釕、乙醇、聚乙烯醇、氨水混合制備得4mg/ml氧化石墨烯混合溶液;其中,氧化石墨烯與貴金屬化合物總量的質量比為1:0.05(氯金酸和三氯化釕的質量比為1:1);乙醇占氧化石墨烯混合溶液的質量百分比為20%,聚乙烯醇占氧化石墨烯混合溶液的質量百分比為1%;氧化石墨烯混合液中,氧化石墨烯與氨的質量比為1:50。(3)將步驟(2)得到的氧化石墨烯混合溶液注入細管狀反應器中,將不封口(空氣氣氛)的輻照反應器用鈷60γ射線源輻照得石墨烯-貴金屬納米顆粒復合水凝膠;其中,劑量為110kGy,輻照反應的劑量率為1kGy/小時。(4)將步驟(3)所得石墨烯-貴金屬納米顆粒復合水凝膠進行超臨界二氧化碳干燥,得細長棒狀石墨烯-鉑納米顆粒復合氣凝膠。該石墨烯-貴金屬納米顆粒復合氣凝膠具有良好的吸油性能,在小分子電分析和電催化等有機反應催化合成領域、石油有機中間體的合成領域等方面均具有巨大的潛在應用價值。其碳氧比、密度和吸附容量如表1所示。其內部微觀結構與圖1和圖2類似。實施例3(1)氧化石墨烯分散液的制備方法與實施例1相同。(2)將步驟(1)制得的氧化石墨烯分散液、氯金酸和甲醇、丙二醇、丁醇和甲胺混合制備得8mg/ml氧化石墨烯混合溶液;其中,氧化石墨烯與氯金酸的質量比為1:0.01;甲醇、丙二醇和丁醇占氧化石墨烯混合溶液的質量百分比均為5%,氧化石墨烯混合液中,氧化石墨烯與甲胺的質量比為1:1。(3)將步驟(2)得到的氧化石墨烯混合溶液注入圓柱狀輻照反應器中,通氬氣除氧;將封好的輻照反應器用鈷60γ射線源輻照得石墨烯-金納米顆粒復合水凝膠;其中,劑量為600kGy,輻照反應的劑量率為10kGy/小時。(4)將步驟(3)所得石墨烯-金納米顆粒復合水凝膠進行冷凍干燥,得干態的圓柱狀石墨烯-金納米顆粒氣凝膠。所得石墨烯-金納米顆粒復合氣凝膠具有良好的吸油性能,在小分子電分析和電催化等有機反應催化合成領域、石油有機中間體的合成領域等方面均具有巨大的潛在應用價值。其碳氧比、密度和吸附容量如表1所示。其內部微觀結構與圖1和圖2類似。實施例4(1)氧化石墨烯分散液的制備方法與實施例1相同。(2)將步驟(1)制得的氧化石墨烯分散液、氯鈀酸、丙醇、丁二醇和乙二胺混合制備得15mg/ml氧化石墨烯混合溶液;其中,氧化石墨烯與氯鈀酸的質量比為1:0.5;丙醇、丁二醇占氧化石墨烯混合溶液的質量百分比均為2.5%;氧化石墨烯混合液中,氧化石墨烯與乙二胺的質量比為1:100。(3)將步驟(2)得到的氧化石墨烯混合溶液注入圓柱狀輻照反應器中,通氮氣除氧;將封好的輻照反應器用鈷60γ射線源輻照得石墨烯-鈀納米顆粒復合水凝膠;其中,劑量為400kGy,輻照反應的劑量率為15kGy/小時。(4)將步驟(3)所得石墨烯-鈀納米顆粒復合水凝膠進行冷凍干燥,得干態的圓柱狀石墨烯-鈀納米顆粒氣凝膠。該石墨烯-鈀納米顆粒復合氣凝膠具有良好的吸油性能,在小分子電分析和電催化等有機反應催化合成領域、石油有機中間體的合成領域等方面均具有巨大的潛在應用價值。其碳氧比、密度和吸附容量如表1所示。其內部微觀結構與圖1和圖2類似。實施例5(1)氧化石墨烯分散液的制備方法與實施例1相同。(2)將步驟(1)制得的氧化石墨烯分散液、氯鈀酸和氯鉑酸的混合物 (氯金酸和氯鉑酸的質量比為1:1)、丙三醇、乙醇和丙二胺、N,N'-二(2-氨乙基)-1,3-丙二胺混合制備得20mg/ml氧化石墨烯混合溶液。其中,氧化石墨烯與貴金屬化合物的總質量比為1:5;丙三醇占氧化石墨烯混合溶液的質量百分比為1%,乙醇占氧化石墨烯混合溶液的質量百分比為40%,氧化石墨烯混合液中,氧化石墨烯與丙二胺的質量比為1:2.5,氧化石墨烯與N,N'-二(2-氨乙基)-1,3-丙二胺的質量比為1:2.5。(3)將步驟(2)得到的氧化石墨烯混合溶液注入圓柱狀輻照反應器中,通氮氣除氧;將封好的輻照反應器用鈷60γ射線源輻照得石墨烯-貴金屬無機納米顆粒復合水凝膠;其中,劑量為500kGy,輻照反應的劑量率為6kGy/小時。(4)將步驟(3)所得石墨烯-貴金屬無機納米顆粒復合水凝膠進行冷凍干燥,得干態的圓柱狀石墨烯-貴金屬無機納米顆粒氣凝膠。該石墨烯-貴金屬無機納米顆粒復合氣凝膠具有良好的吸油性能,在小分子電分析和電催化等有機反應催化合成領域、石油有機中間體的合成領域等方面均具有巨大的潛在應用價值。其碳氧比、密度和吸附容量如表1所示。其內部微觀結構與圖1和圖2類似。實施例6(1)氧化石墨烯分散液的制備方法與實施例1相同。(2)將步驟(1)制得的氧化石墨烯分散液、三氯化銠和三氯化銥的混合物(三氯化銠和三氯化銥的質量比為1:1)、甲醇、異丁醇和二乙烯三胺、三乙烯四胺、四乙烯五胺混合制備得2mg/ml氧化石墨烯混合溶液;其中,氧化石墨烯與貴金屬化合物的總質量比為1:1;氧化石墨烯混合液中,氧化石墨烯與二乙烯三胺的質量比為1:0.5,氧化石墨烯與三乙烯四胺的質量比為1:0.1,氧化石墨烯與四乙烯五胺的質量比為1:0.1;甲醇占氧化石墨烯混合溶液的質量百分比為40%,異丁醇占氧化石墨烯混合溶液的質量百分比為40%。(3)將步驟(2)得到的氧化石墨烯混合溶液注入圓柱狀輻照反應器中, 通氮氣除氧;將封好的輻照反應器用鈷60γ射線源輻照得石墨烯-貴金屬無機納米顆粒復合水凝膠;其中,劑量為500kGy,輻照反應的劑量率為12kGy/小時。(4)將步驟(3)所得石墨烯-貴金屬無機納米顆粒復合水凝膠進行冷凍干燥,得干態的圓柱狀石墨烯-貴金屬無機納米顆粒氣凝膠。該石墨烯-貴金屬無機納米顆粒復合氣凝膠具有良好的吸油性能,在小分子電分析和電催化等有機反應催化合成領域、石油有機中間體的合成領域等方面均具有巨大的潛在應用價值。其碳氧比、密度和吸附容量如表1所示。其內部微觀結構與圖1和圖2類似。實施例7(1)氧化石墨烯分散液的制備方法與實施例1相同。(2)將步驟(1)制得的氧化石墨烯分散液、氯釕酸鉀和乙醇、聚烯丙基胺混合制備得10mg/ml氧化石墨烯混合溶液;其中,氧化石墨烯與氯釕酸鉀的質量比為1:0.5;乙醇占氧化石墨烯混合溶液的質量百分比為90%;氧化石墨烯混合液中,氧化石墨烯與聚烯丙基胺的質量比為1:0.5。(3)將步驟(2)得到的氧化石墨烯混合溶液注入圓柱狀輻照反應器中,通氬氣除氧;將封好的輻照反應器用鈷60γ射線源輻照得石墨烯-釕納米顆粒復合水凝膠;其中,劑量為300kGy,輻照反應的劑量率為15kGy/小時。(4)將步驟(3)所得石墨烯-釕納米顆粒復合水凝膠進行超臨界二氧化碳干燥,得干態的圓柱狀石墨烯-釕納米顆粒氣凝膠。該石墨烯-釕納米顆粒復合氣凝膠具有良好的吸油性能,在小分子電分析和電催化等有機反應催化合成領域、石油有機中間體的合成領域等方面均具有巨大的潛在應用價值。其碳氧比、密度和吸附容量如表1所示。其內部微觀結構與圖1和圖2類似。實施例8(1)氧化石墨烯分散液的制備方法與實施例1相同。(2)將步驟(1)制得的氧化石墨烯分散液、三氯化釕和丙醇混合制備 得15mg/ml氧化石墨烯混合溶液;其中,氧化石墨烯與三氯化釕的質量比為1:0.5,丙醇占氧化石墨烯混合溶液的質量百分比為10%。(3)將氧化石墨烯混合液的扁平狀輻照反應器中,在通氮氣除氧,用電子束射線源輻照,得扁平狀石墨烯-釕納米顆粒復合水凝膠;其中,劑量為600kGy,輻照反應的劑量率為8kGy/小時。(4)將步驟(3)所得石墨烯-釕納米顆粒復合水凝膠進行冷凍干燥,得扁平狀石墨烯-釕納米顆粒復合氣凝膠。該石墨烯-釕納米顆粒復合氣凝膠具有良好的吸油性能,在小分子電分析和電催化等有機反應催化合成領域、石油有機中間體的合成領域等方面均具有巨大的潛在應用價值。其碳氧比、密度和吸附容量如表1所示。其內部微觀結構與圖1和圖2類似。實施例9(1)氧化石墨烯分散液的制備方法與實施例1相同。(2)將步驟(1)制得的氧化石墨烯分散液、氯銠酸和乙二胺混合制備得10mg/ml氧化石墨烯混合溶液;其中,氧化石墨烯與氯銠酸的質量比為1:3;氧化石墨烯混合液中,氧化石墨烯與乙二胺的質量比為1:200。(3)將步驟(2)得到的氧化石墨烯混合溶液注入圓柱狀輻照反應器中,通氮氣除氧;將封好的輻照反應器用鈷60γ射線源輻照得石墨烯-銠納米顆粒復合水凝膠;其中,劑量為800kGy,輻照反應的劑量率為15kGy/小時。(4)將步驟(3)所得石墨烯-銠納米顆粒復合水凝膠進行超臨界二氧化碳干燥,得干態的圓柱狀石墨烯-銠納米顆粒氣凝膠。該石墨烯-銠納米顆粒復合氣凝膠具有良好的吸油性能,在小分子電分析和電催化等有機反應催化合成領域、石油有機中間體的合成領域等方面均具有巨大的潛在應用價值。其碳氧比、密度和吸附容量如表1所示。其內部微觀結構與圖1和圖2類似。實施例10(1)氧化石墨烯分散液的制備方法與實施例1相同。(2)將步驟(1)制得的氧化石墨烯分散液、氯銠酸鉀和乙二胺混合制備得10mg/ml氧化石墨烯混合溶液;其中,氧化石墨烯與氯銠酸鉀的質量比為1:3;氧化石墨烯混合液中,氧化石墨烯與乙二胺的質量比為1:150。(3)將步驟(2)得到的氧化石墨烯混合溶液注入圓柱狀輻照反應器中,通氮氣除氧;將封好的輻照反應器用鈷60γ射線源輻照得石墨烯-銠納米顆粒復合水凝膠;其中,劑量為50kGy,輻照反應的劑量率為0.1kGy/小時。(4)將步驟(3)所得石墨烯-銠納米顆粒復合水凝膠進行超臨界二氧化碳干燥,得干態的圓柱狀石墨烯-銠納米顆粒氣凝膠。該石墨烯-銠納米顆粒復合氣凝膠具有良好的吸油性能,在小分子電分析和電催化等有機反應催化合成領域、石油有機中間體的合成領域等方面均具有巨大的潛在應用價值。其碳氧比、密度和吸附容量如表1所示。其內部微觀結構與圖1和圖2類似。對比例1(1)氧化石墨烯分散液的制備方法與實施例1相同。(2)將步驟(1)制得的氧化石墨烯分散液、氯金酸、丙醇混合制備得4mg/ml氧化石墨烯混合液;其中,丙醇占氧化石墨烯混合溶液的質量百分比為0.5%,氧化石墨烯與氯金酸的質量比為1:0.5。(3)將步驟(2)得到的氧化石墨烯混合溶液注入圓柱狀輻照反應器中,通氮氣除氧;將封好的輻照反應器用鈷60γ射線源輻照;其中,劑量為300kGy,輻照反應的劑量率為3kGy/小時。由于水溶性還原劑的含量比較少,得到的是懸浮在管內的石墨烯-金納米復合顆粒,得不到具有一定自支撐力學強度的連續濕態石墨烯-金納米顆粒復合水凝膠,同樣也得不到連續的干態石墨烯-金納米顆粒復合氣凝膠。其產物不具有良好吸油性能,碳氧比如表1中所示。對比例2(1)氧化石墨烯分散液的制備方法與實施例1相同。(2)將步驟(1)制備的氧化石墨烯分散液與氯釕酸鉀混合制備得2 mg/ml氧化石墨烯混合液;其中,氧化石墨烯與氯釕酸鉀的質量比為1:0.5。(3)將步驟(2)得到的氧化石墨烯混合溶液注入圓柱狀輻照反應器中,通氮氣除氧;將封好的輻照反應器用鈷60γ射線源輻照;其中,劑量為300kGy,輻照反應的劑量率為3kGy/小時。由于無水溶性還原劑存在,其結果得到的是氧化石墨烯與氯釕酸鉀的混合液,得不到連續的塊體石墨烯-釕納米顆粒復合水凝膠,也得不到干態的石墨烯-釕納米顆粒復合氣凝膠。其產物不具有良好吸油性能,碳氧比如表1中所示。對比例3(1)氧化石墨烯分散液的制備方法與實施例1相同。(2)將步驟(1)制備的氧化石墨烯分散液、三氯化銠與丙醇混合制備得4mg/ml氧化石墨烯混合液;其中,丙醇占氧化石墨烯混合溶液的質量百分比為99%,氧化石墨烯與三氯化銠的質量比為1:0.5。(3)將步驟(2)得到的氧化石墨烯混合溶液注入圓柱狀輻照反應器中,通氮氣除氧;將封好的輻照反應器用鈷60γ射線源輻照;其中,劑量為300kGy,輻照反應的劑量率為3kGy/小時。由于水溶性還原劑的含量較多,致使氧化石墨烯以及貴金屬離子在溶液中的分散性較差,其結果是得不到連續的石墨烯-銠納米顆粒復合水凝膠,也得不到連續的干態石墨烯-銠納米顆粒氣凝膠。其產品不具有良好吸油性能,碳氧比如表1中所示。對比例4(1)氧化石墨烯分散液的制備方法與實施例1相同。(2)將步驟(1)制備的氧化石墨烯分散液、氯鈀酸與丙醇混合制備得4mg/ml氧化石墨烯混合液;其中,丙醇占氧化石墨烯混合溶液的質量百分比為30%,氧化石墨烯與氯鈀酸的質量比為1:0.5。(3)將氧化石墨烯混合液裝入輻照管中,通氮氣除氧;靜置24小時不進行輻照。由于沒有進行輻照反應,其結果是得不到連續的石墨烯-鈀納米顆粒復合水凝膠,也得不到連續的干態石墨烯-鈀納米顆粒復合氣凝膠。其產物不具有良好吸油性能,碳氧比如表1中所示。對比例5(1)將氯金酸水溶液與丙醇混合制備得貴金屬離子混合液;其中,丙醇占混合溶液的質量百分比為30%,氯金酸在貴金屬離子混合液的質量濃度為4mg/ml。(2)將貴金屬離子混合液裝入輻照管中,通氮氣除氧;將封好的輻照反應器用鈷60γ射線源輻照;其中,劑量為600kGy,輻照反應的劑量率為10kGy/小時。由于沒有氧化石墨烯的引入,其結果得到的是團聚的肉眼可見(毫米級)金顆粒。效果實施例測定實施例1-10所得石墨烯-貴金屬無機納米顆粒復合氣凝膠和對比例1-5所得產物的碳氧比、氣凝膠密度和吸油性能數據。其中,碳元素含量和氧元素含量通過X射線光電子能譜XPS得到,碳氧比是根據碳元素含量和氧元素含量比值得到;氣凝膠密度根據本領域常規手段測試,由質量與體積比得到;吸油性能根據本領域常規手段測試,其數值由吸油質量與氣凝膠質量比得到,吸附容量以正十烷為例。測試結果見表1。表1實施例及對比例終產品的碳氧比、密度和吸油性能數據樣品碳氧比密度/mg/cm3吸油性能/g/g實施例18.003.0250實施例27.726.2140實施例38.939.070實施例48.1123.235實施例58.231286實施例68.315.7120實施例78.2815.644實施例89.172136實施例99.314318實施例107.283323對比例14.10——對比例23.20——對比例33.92——對比例42.30——對比例5———當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1
- 過渡金屬與石墨烯的復合納米線及其制備方法與流程
- 一種氧化石墨烯表面原位生長二氧化硅的納米雜化填料及其制備方法與流程
- GO增強CNT覆膜砂的智能混凝土無線傳感器的制造方法與工藝
- 一種基于納米纖維素晶須MoS2/石墨烯復合對電極材料及其制備方法和應用與流程
- 聚醚砜樹脂/石墨烯納米片多孔納米復合材料、制備方法及其用途與流程
- 一種改性石墨烯與納米纖維素復合溫敏材料的制備方法與制造工藝
- 石墨烯摻雜改性納米鈣鈦礦型La1?xSrxMnO3復合材料及其制備方法和應用與制造工藝
- 一種在二氧化錳電極表面對苯胺進行原位氧化的方法與制造工藝
- 一種氧化石墨烯/銀納米顆粒/金字塔形硅三維拉曼增強基底及制備方法和應用與制造工藝
- 納米羧基化石墨烯復合滌綸導電織物的制備方法與制造工藝