一種耐硫甲烷化催化劑及其制備方法以及氫氣與CO甲烷化的方法與流程
本發明涉及一種耐硫甲烷化催化劑及其制備方法以及氫氣與CO甲烷化的方法,具體地,涉及一種含有納米級的Zr(MoO4)2和ZrO2,以及將含有鉬源、鋯源和固體有機胺的固體混合物與水蒸汽汽固相接觸從而發生反應制備該催化劑的方法,以及在該催化劑存在下將氫氣與CO進行反應得到甲烷的方法。
背景技術:
甲烷化反應是指合成氣中CO在一定的溫度、壓力以及催化劑的作用下與H2進行反應生成甲烷的過程。其反應式可表示如下:
CO+3H2=CH4+H2O (1)
CO+H2O=CO2+H2 (2)
2CO+2H2=CH4+CO2 (3)
通過甲烷化反應將經煤氣化得到的合成氣轉化為甲烷,是煤碳潔凈利用的最佳方案之一,該過程被稱為煤制天然氣。
甲烷化反應作為煤制天然氣的重要工序,分為完全甲烷化和直接甲烷化兩種方式。
完全甲烷化一般采用鎳基催化劑,由于鎳很容易與原料氣中的硫化物反應而引起中毒失活,需要在甲烷化工序前,利用低溫甲醇洗技術脫除原料氣中的H2S等含硫化合物。由于低溫甲醇洗的操作溫度為-40℃,而甲烷化反應器的入口溫度為300℃,因此原料氣要經過先降溫后升溫的過程,能量效率低。
直接甲烷化一般采用耐硫的甲烷化催化劑,該過程中低溫甲醇洗工序放 在甲烷化工序之后,可以有效回避原料氣降溫升溫過程中的能量損失。耐硫甲烷化催化劑一般采用鉬基催化劑,負載于Al2O3、ZrO2等載體上。
US4151191公開了一種由含H2、CO和硫化物的氣體混合物生產CH4或含CH4氣體的方法,使用的甲烷化催化劑包括:鑭系和/或錒系金屬氧化物以及Mo金屬氧化物,其中鑭系和/或錒系金屬與Mo的原子比為9:1。該催化劑在H2/CO為1:1和硫化物含量高達3重量%的條件下表現出極較好的甲烷化催化特性。
US4320030公開了一種用于甲烷化反應的催化劑,該催化劑包括:含有Mo、V和/或W的化合物混合物、或者Mo、V和W中兩種或更多種的化合物混合物。該催化劑的制備方法如下:先將其活性與穩定劑等組分前體與固體硫或硫化物混合,然后在惰性氣氛或H2S/H2氣氛下對所述固體進行煅燒和冷卻,最后用稀釋的含氧氣流鈍化所述催化劑,并進行粉碎、研磨和造粒,最終形成所要求的催化劑。
CN103157485A公開了一種負載型的耐硫甲烷化催化劑,包括:0-20份(重量)催化劑助劑(M1)AOB;5-90份(重量)催化劑活性組分(M2)COD;5-90份(重量)載體改性劑(M3)EOF和100份(重量)多孔載體(M4)GOH,其中M1為Co、Ni、La和/或K;M2為Mo、W和/或V;M3為Ce、Zr、Ti、Mg和/或Si;M4為Ce或Al,并且M3與M4不相同;(M3)EOF和(M4)GOH也可被ZrO2、TiO2、MgO和/或SiO2所取代。
采用耐硫甲烷化催化劑的直接甲烷化工序為多段固定床串聯工藝。隨著甲烷化反應的持續進行,反應體系中產生的CH4和CO2含量逐漸升高。在末段甲烷化反應器中,高濃度CH4和CO2會抑制甲烷化反應式(3)的進行,限制H2和CO進一步轉化為CH4和CO2。此時,要求在末段甲烷化反應器中使用具有更高甲烷化反應催化活性的催化劑。然而,現有大多數以Al2O3為載體的甲烷化催化劑達不到上述要求。
CN103962123A公布了一種采用ZrO2為載體的鉬基催化劑,活性與Al2O3載體相比有較大提高,但工業仍需要活性更好的催化劑,且該發明公開的催化劑制備繁瑣,成本較高。
技術實現要素:
本發明的目的是為了提高甲烷化反應中CO的轉化率,提供高活性的催化劑,且該催化劑制備方法簡單,可以有效降低生產成本,提供了一種耐硫甲烷化催化劑及其制備方法以及氫氣與CO甲烷化的方法。
為了實現上述目的,本發明提供了一種耐硫甲烷化催化劑,該催化劑含有納米Zr(MoO4)2顆粒和納米ZrO2顆粒,其中,ZrO2的晶相為單斜相。
本發明還提供了一種制備本發明提供的甲烷化催化劑的方法,該方法包括:(1)將含有鉬源、鋯源和固體有機胺的固體混合物與水蒸汽在密閉容器中接觸,得到鉬酸鋯和氫氧化鋯的混合物;水蒸汽的溫度為100-250℃;(2)將步驟(1)中所述接觸得到的混合物進行干燥和焙燒,得到含有Zr(MoO4)2和ZrO2的催化劑。
本發明還提供了一種本發明提供的方法制備得到的甲烷化催化劑。
本發明還提供了一種氫氣與CO甲烷化的方法,該方法包括:在甲烷化條件下,將氫氣與CO在甲烷化催化劑存在下反應,得到甲烷;其中,所述甲烷化催化劑為本發明提供的催化劑。
本發明提供的甲烷化催化劑含有納米級的Zr(MoO4)2和ZrO2。其中ZrO2作為載體,晶相為單斜相;Zr(MoO4)2為活性組分,可以在氫氣與CO甲烷化的反應中獲得更高的CO的轉化率,有更好的催化活性。
本發明提供的制備含有Zr(MoO4)2和ZrO2的甲烷化催化劑方法中,采用將水蒸汽與含有鉬源、鋯源和固體有機胺的固體反應物在密閉容器中接觸,不僅不需要反應產物分離,而且可以改善獲得的催化劑的孔結構,并直接獲 得活性組分與載體的復合,簡化了催化劑制備過程而且水消耗小,減少廢水處理。
本發明提供的甲烷化催化劑的催化活性更高,反應活性高于負載型的MoO3/ZrO2催化劑。同時制備方法簡單,為汽固相一步反應過程,沒有產品的分離問題,有效節省催化劑生產成本。
本發明的其它特征和優點將在隨后的具體實施方式部分予以詳細說明。
附圖說明
附圖是用來提供對本發明的進一步理解,并且構成說明書的一部分,與下面的具體實施方式一起用于解釋本發明,但并不構成對本發明的限制。在附圖中:
圖1為本發明一種優選實施方式的實施示意圖;
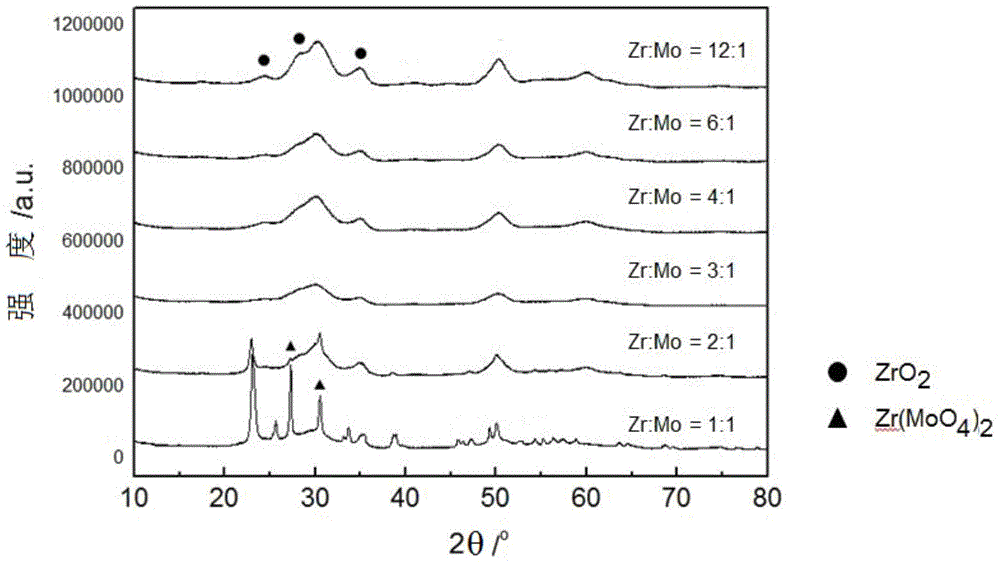
圖2為制備的甲烷化催化劑的XRD譜圖;
圖3為實施例3制備的甲烷化催化劑的透射電鏡照片;
圖4為各催化劑進行50h甲烷化反應的轉化率。
附圖標記說明
1、壓力容彈 2、反應物料 3、多孔篩網 4、水
具體實施方式
以下對本發明的具體實施方式進行詳細說明。應當理解的是,此處所描述的具體實施方式僅用于說明和解釋本發明,并不用于限制本發明。
本發明提供了一種耐硫甲烷化催化劑,該催化劑含有納米Zr(MoO4)2顆粒和納米ZrO2顆粒,其中,ZrO2的晶相為單斜相。
根據本發明,催化劑的平均粒徑為5-10nm。其中含有的Zr(MoO4)2顆粒和ZrO2顆粒雖然無法直接測定,但是也都平均粒徑小于催化劑的平均粒徑, 為納米級顆粒。可以通過透射電鏡方法采用JEOL公司JEM-ARM200F型號的透射電子顯微鏡測定粒徑。圖3中顯示了實施例3制備的甲烷化催化劑的透射電鏡照片,可以測定催化劑的平均粒徑。其他實施例制備的甲烷化催化劑的透射電鏡照片相似,本發明中不一一顯示,但列出測定的催化劑平均粒徑。
根據本發明,該催化劑的BET比表面積為180-250m2/g,孔體積為0.15-0.25cm3/g。
根據本發明,可以通過XRD譜圖分析,如圖2所示,確定所述催化劑中含有Zr(MoO4)2和ZrO2。所述催化劑的XRD譜圖中,在2θ為27.2°、30.5°處有Zr(MoO4)2的特征峰,在2θ為24.7°、28.4°、34.9°處有ZrO2的特征峰。
根據本發明,提供的甲烷化催化劑中,Zr(MoO4)2作為活性組分,ZrO2作為載體。優選情況下,以該催化劑的總重量為基準,Zr(MoO4)2的含量為10-40重量%,ZrO2的含量為60-90重量%。可以通過XRF分析確定所述催化劑中,Zr(MoO4)2和ZrO2的含量。
本發明中,可以進一步優選地,所述催化劑中,Zr與Mo的摩爾比為3:1至12:1。催化劑組分在該限定范圍內可以有更好的反應催化活性。
本發明還提供了一種制備本發明提供的甲烷化催化劑的方法,該方法包括:(1)將含有鉬源、鋯源和固體有機胺的固體混合物與水蒸汽在密閉容器中接觸,得到鉬酸鋯和氫氧化鋯的混合物;水蒸汽的溫度為100-250℃;(2)將步驟(1)中所述接觸得到的混合物進行干燥和焙燒,得到含有Zr(MoO4)2和ZrO2的催化劑。
根據本發明,步驟(1)中,所述鉬源、鋯源與所述固體有機胺的用量只要滿足制備所述甲烷化催化劑中含有足夠的Zr(MoO4)2和ZrO2的含量即可。優選情況下,所述鋯源中元素Zr和所述鉬源中元素Mo的總摩爾量: 固體有機胺的摩爾量為1:2-10。
根據本發明,步驟(1)中,加入的所述鉬源與鋯源的用量滿足得到的甲烷化催化劑中,Zr(MoO4)2和ZrO2的含量的即可。優選情況下,所述鋯源中元素Zr和所述鉬源中元素Mo的摩爾比為3-15:1。
根據本發明,在步驟(1)中的水蒸汽的溫度為100-250℃條件下,水蒸汽與所述固體混合物進行汽固相接觸并發生反應,形成鉬酸鋯和二氧化鋯的前驅體(即氫氧化鋯)。本發明保證與所述固體混合物進行接觸的水蒸汽的量,能夠完全使鉬源與一部分鋯源轉變為鉬酸鋯,另一部分鋯源轉變為氫氧化鋯。優選地,所述固體混合物與水的摩爾比不大于1:10,且不小于1:100。
根據本發明,所述鋯源沒有特別地限定,優選情況下,所述鋯源為在步驟(1)中所述接觸的條件下能夠轉變為氫氧化鋯的物質;優選地,所述鋯源為硝酸氧鋯、氧氯化鋯、碳酸鋯和硝酸鋯中的至少一種。
根據發明,優選情況下,所述鉬源為在步驟(1)中所述接觸的條件下能夠轉變為鉬酸鋯的物質;優選地,所述鉬源為鉬酸銨和/或鉬酸鈉。
根據本發明,所述固體有機胺沒有特別地限定,優選情況下,所述固體有機胺為尿素和/或六次甲基四胺。
本發明中,含有所述鉬源、鋯源和固體有機胺的固體混合物的顆粒直徑可以為150-300μm。
本發明方法的整個接觸過程中沒有液相參與。
本發明提供的方法在密閉容器中實施,其中接觸的壓力為對應步驟(1)中的水蒸汽的溫度的水的飽和蒸汽壓。
本發明中,水的用量為足以在步驟(1)進行的所述接觸中生成飽和蒸汽。
根據本發明,步驟(1)中所述接觸的條件以及步驟(2)中干燥和焙燒的條件沒有特別的限定。優選情況下,所述接觸的時間為1-50h;所述干燥 的溫度為100-120℃,所述干燥的時間為10-14h,所述焙燒的溫度為400-600℃,所述焙燒的時間為2-6h。
根據本發明,一種優選的制備所述甲烷化催化劑的方法具體為:如圖1所示,所述密閉容器包括多孔篩網,將含有鉬源、鋯源和固體有機胺的固體混合物與水蒸汽接觸的方式包括在密閉容器內填充水,將含有鉬源、鋯源和固體有機胺的固體混合物置于多孔篩網上,多孔篩網位于水面上且多孔篩網的下表面與水面之間有間隙,然后加熱,使密閉容器內的水形成水蒸汽,水蒸汽穿過多孔篩網,到達位于多孔篩網上的含有鋯源和固體有機胺的固體混合物表面。所述密閉容器可以為壓力容彈。
根據本發明,所述多孔篩網可以沒有特別地限定,多孔篩網的孔徑大于固體混合物的顆粒直徑,不使固體混合物下落即可。優選情況下,所述多孔篩網的孔隙率為4000-250000孔/cm2,所述多孔篩網的孔徑為20-150μm。
本發明還提供了一種本發明提供的方法制備得到的甲烷化催化劑。
所述甲烷化催化劑具有上述特征,在此不再贅述。該甲烷化催化劑中Zr(MoO4)2和ZrO2的含量,以及元素Zr與Mo的摩爾比可以通過X-射線熒光光譜法測定得到。
本發明還提供了一種氫氣與CO甲烷化的方法,該方法包括:在甲烷化條件下,將氫氣與CO在甲烷化催化劑存在下反應,得到甲烷;其中,所述甲烷化催化劑為本發明提供的催化劑。
所述H2與CO直接甲烷化的條件為,先將催化劑在400-500℃下以含有3體積%的H2S的氫氣氣氛(50ml/gcat/min)進行硫化2-5h;然后將H2、H2S和CO的混合氣(其中,H2/CO體積比為1:1至3:1,H2S的濃度大于0.1體積%)在1-4MPa、400-500℃下,以體積空速4000-10000h-1與催化劑接觸反應。
以下將通過實施例對本發明進行詳細描述。
以下實施例中甲烷化催化劑產品通過透射電鏡方法采用JEOL公司JEM-ARM200F型號的透射電子顯微鏡測定粒徑;
通過X射線粉末衍射方法采用Rigaku公司型號為D/max-2600/pc的X-射線衍射儀測定產品的XRD譜圖,與標準譜圖(PDF2-2004)比對,確定為Zr(MoO4)2和單斜相ZrO2;
通過X-射線熒光光譜分析(XRF)采用Rigaku公司型號為ZSX Primus II的X-射線熒光光譜儀上測定甲烷化催化劑中的Zr(MoO4)2和ZrO2含量;
通過氮氣吸附法采用Micromeritics公司tristarII 3020-M型號的比表面分析儀儀器測定產品的孔結構;
CO的轉化率按下式計算:
CO的濃度通過采用BROOKS公司型號為5850E的質量流量計測定。
對比例1
浸漬法制備MoO3/ZrO2。
稱取單斜相氧化鋯(Alfa Aesar購買)載體100g,比表面積為90m2/g。放入110℃干燥箱中烘干12小時,最后在600℃馬弗爐中焙燒4小時;
將11.9g的(NH4)6Mo7O24·4H2O溶解于70g的去離子水中,經攪拌配置成溶液;
將含有(NH4)6Mo7O24的溶液浸漬于上述單斜相氧化鋯載體上,放入110℃干燥箱中烘干12小時,最后在600℃馬弗爐中焙燒4小時,制得催化劑MoO3/ZrO2,記為DC-1,MoO3含量為8.9重量%,ZrO2的含量為91.1重量%;其中Zr:Mo(mol,下同)=12:1。
實施例1
本實施例用于說明本發明提供的制備甲烷化催化劑的方法。
如圖1所示,在壓力容彈中裝入20g的水,將15g的ZrO(NO3)2·2H2O(顆粒直徑為150μm)、3.4g的(NH4)6Mo7O24·4H2O(顆粒直徑為150μm)、9g的尿素(顆粒直徑為300μm)混合均勻,放于篩網上(孔隙率為4000孔/cm2,孔徑為150μm),置于水面以上,不接觸水面,其中尿素:(Zr+Mo)(mol,下同)=2:1,Zr:Mo=3:1。
將壓力容彈密閉加熱,在150℃下保持50h。取出篩網上的產物粉末,在120℃下干燥12h,在500℃下焙燒4h,得到甲烷化催化劑MC-1。
測定MC-1的粒徑為5nm;
測定MC-1的XRD譜圖(圖2)與標準譜圖比對確定為Zr(MoO4)2和ZrO2晶體的混合相;Zr(MoO4)2含量為40重量%和ZrO2的含量為60重量%;
測定MC-1的孔結構,比表面積為220m2/g,孔體積為0.22cm3/g。
實施例2
本實施例用于說明本發明提供的制備甲烷化催化劑的方法。
如圖1所示,在壓力容彈中裝入20g的水,將15g的ZrO(NO3)2·2H2O、2.55g的(NH4)6Mo7O24·4H2O、8.4g的尿素混合均勻,放于篩網上(孔隙率為4000孔/cm2,孔徑為150μm),置于水面以上,不接觸水面,其中尿素:(Zr+Mo)=2:1,Zr:Mo=4:1。
將壓力容彈密閉加熱,在150℃下保持50h。取出篩網上的產物粉末,在120℃下干燥12h,在500℃下焙燒4h,得到甲烷化催化劑MC-2。
測定MC-2的粒徑為5nm;
測定MC-2的XRD譜圖(圖2)與標準譜圖比對確定為Zr(MoO4)2和ZrO2晶體的混合相;Zr(MoO4)2含量為32.3重量%和ZrO2的含量為67.7%重 量%;
測定MC-2的孔結構,比表面積為225m2/g,孔體積為0.23cm3/g。
實施例3
本實施例用于說明本發明提供的制備甲烷化催化劑的方法。
如圖1所示,在壓力容彈中裝入20g的水,將15g的ZrO(NO3)2·2H2O、1.7g的(NH4)6Mo7O24·4H2O、7.88g的尿素混合均勻,放于篩網上(孔隙率為4000孔/cm2,孔徑為150μm),置于水面以上,不接觸水面,其中尿素:(Zr+Mo)=2:1,Zr:Mo=6:1。
將壓力容彈密閉加熱,在150℃下保持50h。取出篩網上的產物粉末,在120℃下干燥12h,在500℃下焙燒4h,得到甲烷化催化劑MC-3。
將MC-3進行透射電鏡分析,如圖3所示,測定MC-3的粒徑為5nm;
測定MC-3的XRD譜圖(圖2)與標準譜圖比對確定為Zr(MoO4)2和ZrO2晶體的混合相;Zr(MoO4)2含量為23.3%和ZrO2的含量為76.7%;
測定MC-3的孔結構,比表面積為228m2/g,孔體積為0.24cm3/g。
實施例4
本實施例用于說明本發明提供的制備甲烷化催化劑的方法。
如圖1所示,在壓力容彈中裝入20g的水,將15g的ZrO(NO3)2·2H2O、0.85g的(NH4)6Mo7O24·4H2O、7.28g的尿素混合均勻,放于篩網上(孔隙率為4000孔/cm2,孔徑為150μm),置于水面以上,不接觸水面,其中尿素:(Zr+Mo)=2:1,Zr:Mo=12:1。
將壓力容彈密閉加熱,在150℃下保持50h。取出篩網上的產物粉末,在120℃下干燥12h,在500℃下焙燒4h,得到甲烷化催化劑MC-4。
測定MC-4的粒徑為5nm;
測定MC-4的XRD譜圖(圖2)與標準譜圖比對確定為Zr(MoO4)2和ZrO2晶體的混合相;Zr(MoO4)2含量為12.7重量%和ZrO2的含量為87.3重量%;
測定MC-4的孔結構,比表面積為230m2/g,孔體積為0.25cm3/g。
實施例5
本實施例用于說明本發明提供的制備甲烷化催化劑的方法。
如圖1所示,在壓力容彈中裝入20g的水,將15g的ZrOCl2·8H2O、1.17g的(NH4)6Mo7O24·4H2O、16g的尿素混合均勻,放于篩網上(孔隙率為4000孔/cm2,孔徑為150μm),置于水面以上,不接觸水面,其中尿素:(Zr+Mo)=5:1,Zr:Mo=7:1。
將壓力容彈密閉加熱,在250℃下保持1h。取出篩網上的產物粉末,在120℃下干燥12h,在600℃下焙燒4h,得到甲烷化催化劑MC-5。
測定MC-5的粒徑為5nm;
測定MC-5的XRD譜圖與標準譜圖比對確定為Zr(MoO4)2和ZrO2晶體的混合相;Zr(MoO4)2含量為20.4重量%和ZrO2的含量為重量79.6%;
測定MC-5的孔結構,比表面積為195m2/g,孔體積為0.19cm3/g。
實施例6
本實施例用于說明本發明提供的制備甲烷化催化劑的方法。
如圖1所示,在壓力容彈中裝入20g的水,將15g的ZrO(NO3)2·2H2O、1.9g的Na2MoO4·2H2O、44.7g的六次甲基四胺混合均勻,放于篩網上(孔隙率為4000孔/cm2,孔徑為150μm),置于水面以上,不接觸水面,其中六次甲基四胺:(Zr+Mo)=5:1,Zr:Mo=7:1。
將壓力容彈密閉加熱,在200℃下保持12h。取出篩網上的產物粉末, 在120℃下干燥12h,在500℃下焙燒4h,得到甲烷化催化劑MC-6。
測定MC-6的粒徑為8nm;
測定MC-6的XRD譜圖與標準譜圖比對確定為Zr(MoO4)2和ZrO2晶體的混合相;Zr(MoO4)2含量為20.4重量%和ZrO2的含量為79.6重量%;
測定MC-6的孔結構,比表面積為215m2/g,孔體積為0.20cm3/g。
實施例7
本實施例用于說明本發明提供的制備甲烷化催化劑的方法。
如圖1所示,在壓力容彈中裝入30g的水,將15g的ZrOCl2、1.0g的(NH4)6Mo7O24·4H2O、53.8g的尿素混合均勻,放于篩網上(孔隙率為4000孔/cm2,孔徑為150μm),置于水面以上,不接觸水面,其中尿素:(Zr+Mo)=10:1,Zr:Mo=15:1。
將壓力容彈密閉加熱,在100℃下保持50h。取出篩網上的產物粉末,在120℃下干燥12h,在400℃下焙燒4h,得到甲烷化催化劑MC-7。
測定MC-7的粒徑為5nm;
測定MC-7的XRD譜圖與標準譜圖比對確定為Zr(MoO4)2和ZrO2晶體的混合相;Zr(MoO4)2含量為10.3重量%和ZrO2的含量為89.7重量%;
測定MC-7的孔結構,比表面積為221m2/g,孔體積為0.23cm3/g。
實施例8
本實施例用于說明本發明提供的制備甲烷化催化劑的方法。
如圖1所示,在壓力容彈中裝入20g的水,將15g的ZrO(NO3)2·2H2O、10.2g的(NH4)6Mo7O24·4H2O、13.5g的尿素混合均勻,放于篩網上(孔隙率為4000孔/cm2,孔徑為150μm),置于水面以上,不接觸水面,其中尿素:(Zr+Mo)=2:1,Zr:Mo=1:1。
將壓力容彈密閉加熱,在150℃下保持50h。取出篩網上的產物粉末,在120℃下干燥12h,在500℃下焙燒4h,得到甲烷化催化劑MC-8。
測定MC-8的粒徑為7nm;
測定MC-8的XRD譜圖(圖2)與標準譜圖比對確定為Zr(MoO4)2和ZrO2晶體的混合相;Zr(MoO4)2含量為77重量%和ZrO2的含量為23重量%;
測定MC-8的孔結構,比表面積為210m2/g,孔體積為0.2cm3/g。
實施例9
本實施例用于說明本發明提供的制備甲烷化催化劑的方法。
如圖1所示,在壓力容彈中裝入20g的水,將15g的ZrO(NO3)2·2H2O、5.1g的(NH4)6Mo7O24·4H2O、10g的尿素混合均勻,放于篩網上(孔隙率為4000孔/cm2,孔徑為150μm),置于水面以上,不接觸水面,其中尿素:(Zr+Mo)=2:1,Zr:Mo=2:1。
將壓力容彈密閉加熱,在150℃下保持50h。取出篩網上的產物粉末,在120℃下干燥12h,在500℃下焙燒4h,得到甲烷化催化劑MC-9。
測定MC-9的粒徑為8nm;
測定MC-9的XRD譜圖(圖2)與標準譜圖比對確定為Zr(MoO4)2和ZrO2晶體的混合相;Zr(MoO4)2含量為52.7重量%和ZrO2的含量為47.3重量%;
測定MC-9的孔結構,比表面積為218m2/g,孔體積為0.21cm3/g。
測試例
將對比例1以及實施例1、2、3、4、8、9所制備的催化劑DC-1、MC-1、MC-2、MC-3、MC-4、MC-8至MC-9分別進行的H2與CO甲烷化反應。
催化劑填裝量為1g,使用含有3體積%H2S的H2氣氛(流量為50ml/min) 硫化催化劑,溫度為450℃,時間為3h;再通入反應氣體H2、H2S和CO的混合氣(其中H2S的濃度為1體積%,H2/CO(體積比)=1:1),混合氣體積空速為5000h-1,反應壓力為3.0MPa,反應溫度為450℃,進行甲烷化反應。
在反應器出口測量產物中H2和CO的濃度,計算進行甲烷化反應50h時CO的轉化率,結果見圖4。
由上述實施例可以看出,本發明提供的甲烷化催化劑含有納米級Zr(MoO4)2和ZrO2,在進行耐硫甲烷化反應的測試例中,表現出優異的活性。從圖4可以看出尤其當催化劑中Zr:Mo=3-12時,CO轉化率均超過對比例1中使用浸漬法制備的DC-1催化劑。本發明提供了催化性能更高的耐硫甲烷化催化劑。
本發明所提供的催化劑的制備方法可以直接得到納米級產物粉末,無需傳統的催化劑制備方法(涉及液相反應)所必須的將固體產物從液相中分離的問題,簡化步驟,減小水的消耗和廢水處理。
- 還沒有人留言評論。精彩留言會獲得點贊!