氮摻雜石墨烯/碳化硅微球納米復合材料及其制備方法和應用與流程
本發明涉及一種選擇性氧化催化劑及其制備方法和應用,具體涉及一種氮摻雜石墨烯/碳化硅微球納米復合材料及其制備方法和應用。
背景技術:
氮摻雜石墨烯是一種新興的可替代貴金屬催化劑的高效選擇性氧化催化劑。在實驗室中得到的氮摻雜石墨烯,其催化效率與已大規模使用的貴金屬催化劑幾乎相當。出于環境保護和降低成本的考慮,氮摻雜石墨烯的制備方法通常采用以下兩種:一種是首先制備石墨烯,然后通過表面活化和化學反應將氮元素摻入石墨烯;另一種是在制備過程中,通過在原料中加入含有氮元素的物質,直接制備得到含有氮元素的石墨烯。上述方法制備得到的催化劑雖然在實驗室中可以選擇性催化一系列化學反應,但這些氮摻雜石墨烯存在催化反應易團聚沉降的缺點。氮摻雜石墨烯僅有一層或數層碳原子厚度,擁有巨大的比表面積,在溶液中易于堆疊、團聚沉降,降低比表面積,導致催化效率大幅下降。另外,在催化反應結束后,需要將催化劑與反應液分離,在分離的過程中,對催化劑的濃縮和堆疊將是不可避免的,這對氮摻雜石墨烯的循環使用造成了障礙。因此,氮摻雜石墨烯在實際應用中仍然存在分散和循環使用方面的問題。
技術實現要素:
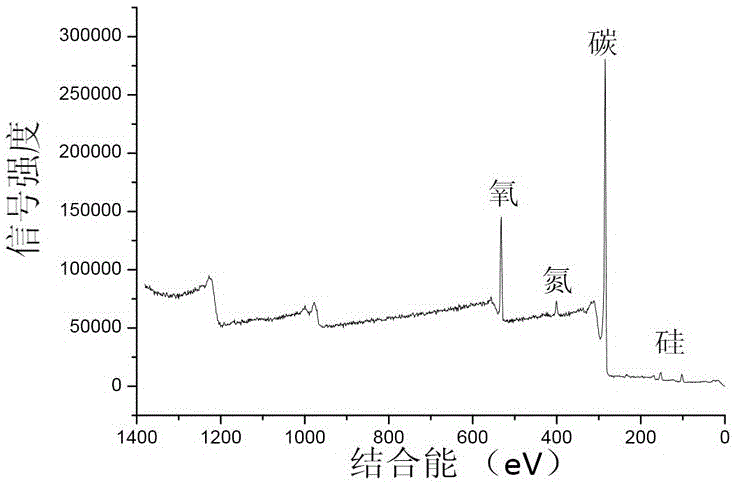
本發明要解決的技術問題是克服現有技術的不足,提供一種抗團聚、耐腐蝕、高催化活性、可多次循環使用的氮摻雜石墨烯/碳化硅微球納米復合材料及其制備方法和應用。為解決上述技術問題,本發明采用的技術方案為一種氮摻雜石墨烯/碳化硅微球納米復合材料,所述氮摻雜石墨烯/碳化硅微球納米復合材料包括碳化硅微球和直立生長于碳化硅微球表面的氮摻雜石墨烯。上述的氮摻雜石墨烯/碳化硅微球納米復合材料中,優選的,所述碳化硅微球為實心結構,所述碳化硅微球的直徑為0.5μm~1.0μm;所述氮摻雜石墨烯的長度為0.1μm~0.3μm。作為一個總的技術構思,本發明還提供一種上述氮摻雜石墨烯/碳化硅微球納米復合材料的制備方法,包括以下步驟:(1)以石墨片或硅片為生長基底,將液態聚硅烷在惰性氣氛中進行高溫裂解,得到負載石墨烯的碳化硅微球;(2)將負載石墨烯的碳化硅微球進行強氧化性酸活化和水合肼還原,然后在惰性氣氛中于高溫下進行保溫,得到氮摻雜石墨烯/碳化硅微球納米復合材料。上述的制備方法中,優選的,所述步驟(1)中,所述高溫裂解的溫度為1300℃~1400℃。上述的制備方法中,優選的,所述步驟(2)的具體操作過程為:將負載石墨烯的碳化硅微球加入強氧化性酸中進行活化處理,經超聲、攪拌、離心、洗滌至中性,得到負載活化石墨烯的碳化硅微球;向所述負載活化石墨烯的碳化硅微球中加入水合肼進行還原處理,然后進行超聲、離心、洗滌和烘干處理,所得產物在惰性氣氛中于900℃~950℃下保溫2h~3h,得到氮摻雜石墨烯/碳化硅微球納米復合材料。上述的制備方法中,優選的,所述負載石墨烯的碳化硅微球與所述強氧化性酸的質量體積比為10mg~20mg∶1mL~2mL,所述強氧化性酸包括濃硫酸與濃硝酸的混合酸或者濃硝酸,所述混合酸中濃硫酸與濃硝酸的質量比為1∶1;所述負載活化石墨烯的碳化硅微球與所述水合肼的質量體積比為10mg~20mg∶1mL~2mL。作為一個總的技術構思,本發明還提供一種上述氮摻雜石墨烯/碳化硅微球納米復合材料的制備方法,包括以下步驟:以石墨片或硅片為生長基底,將液態聚硅烷與液態含氮碳氫化合物的混合物在惰性氣氛中進行高溫裂解,得到氮摻雜石墨烯/碳化硅微球納米復合材料。上述的制備方法中,優選的,所述液態聚硅烷與所述液態含氮碳氫化合物的質量比為100∶3~30;所述液態含氮碳氫化合物包括乙腈或丙腈;所述高溫裂解的溫度為1300℃~1400℃。作為一個總的技術構思,本發明還提供一種上述氮摻雜石墨烯/碳化硅微球納米復合材料或上述制備方法制得的氮摻雜石墨烯/碳化硅微球納米復合材料在催化反應中的應用,包括以下步驟:(1)向氮摻雜石墨烯/碳化硅微球納米復合材料中加入FeCl3與FeCl2的混合溶液和氨水進行磁化,經超聲分散和磁性分離后,得到磁化的氮摻雜石墨烯/碳化硅微球納米復合材料;(2)將去離子水(即反應媒介)、叔丁基過氧化氫(即氧化劑)和還原劑(即待氧化原料)依次加入到磁化的氮摻雜石墨烯/碳化硅微球納米復合材料中,密封進行選擇性氧化催化反應,完成處理。上述的應用中,優選的,所述步驟(1)中,所述氮摻雜石墨烯/碳化硅微球納米復合材料、FeCl3與FeCl2的混合溶液、氨水的用量比值為20mg∶1mL∶0.01mL~0.1mL,所述FeCl3與FeCl2的混合溶液的摩爾濃度為10mmol/L~20mmol/L,FeCl3與FeCl2的摩爾比為1∶1;所述步驟(2)中,所述去離子水、叔丁基過氧化氫、還原劑和磁化的氮摻雜石墨烯/碳化硅微球納米復合材料的用量比值為1mL~3mL∶150μL~200μL∶35μL~80μL∶20mg,所述還原劑包括苯甲醇及其衍生物、乙基苯及其衍生物中的一種或多種,所述選擇性氧化催化反應的溫度為70℃~90℃,反應時間為24h~26h。本發明中,由于氮摻雜石墨烯直立生長于碳化硅微球表面,因此氮摻雜石墨烯層的厚度可認為是氮摻雜石墨烯的長度。本發明的制備方法中,惰性氣氛通常優選為N2或Ar。本發明制備的氮摻雜石墨烯/碳化硅微球納米復合材料中氮的原子百分比含量通常在2at%~3at%范圍內。本發明的制備方法中,由于微球的制備過程是一個有機-無機轉變過程,隨著有機球體在高溫下的收縮、結晶過程,導致微球的外形并非全為標準球體,部分球出現了形變,成為橢球體,或過度的接觸熔融導致新生成的微球并非標準球體。本發明的應用中,催化反應(主要是選擇性氧化催化反應)可催化一系列含C-H鍵的大量有機物,以苯甲醇和乙苯為例,本發明的催化劑對苯環上多種烷基取代物均可進行有效催化。與現有技術相比,本發明的優點在于:1.本發明的氮摻雜石墨烯/碳化硅微球納米復合材料為一種選擇性氧化催化劑,包括實心碳化硅微球和直立生長于微球表面的氮摻雜石墨烯。該結構的生長過程以液態聚硅烷為微液滴,經過高溫固化,液滴發生裂解交聯收縮而沉降在生長基底上,由于微球內部發生碳化硅結晶過程,導致微球內多余的碳向表面富集,最終在高溫下生成了石墨微晶。而微球裂解產生的含碳氣體使得石墨微晶迅速生長成為多層石墨烯,經過該過程,得到的石墨烯牢固的生長在微球表面,這種直立生長的多層石墨烯在后續應用中不會發生團聚而使實際比表面積嚴重降低。使用其他方法制備的石墨烯因沒有合適的載體,難以在實際應用中保持單分散狀態,一旦發生團聚就難以再次分散,使實際參與反應的石墨烯的比表面積嚴重下降。2.本發明的制備方法以石墨片或硅片為生長基底,石墨片或硅片與聚硅烷浸潤性較差,可使聚硅烷裂解產生的液滴(聚硅烷揮發裂解后產生的氣體包含大量微液滴)在生長基底上保持球形外觀,而在其他浸潤性較好的生長基底上則會發生聚硅烷的鋪展,不能得到產物的球狀外觀。本發明僅通過一次高溫裂解過程,便可制備直立生長于碳化硅微球表面的多層石墨烯,即同時制備了多層石墨烯和石墨烯載體(實心碳化硅微球),并使二者牢固結合。3.本發明的氮摻雜石墨烯/碳化硅微球納米復合材料可廣泛應用于高效能選擇性有機催化反應領域。附圖說明圖1為本發明實施例1中制備氮摻雜石墨烯/碳化硅微球納米復合材料的裝置示意圖。圖2為本發明實施例1中氮摻雜石墨烯/碳化硅微球納米復合材料的X光電子能譜圖。圖3為本發明實施例1中氮摻雜石墨烯/碳化硅微球納米復合材料的掃描電子顯微鏡圖(放大倍數為60000倍)。圖4為本發明實施例1中氮摻雜石墨烯/碳化硅微球納米復合材料的掃描電子顯微鏡圖(放大倍數為10000倍)。圖5為本發明實施例1中氮摻雜石墨烯/碳化硅微球納米復合材料的透射電子顯微鏡圖。具體實施方式以下結合說明書附圖和具體優選的實施例對本發明作進一步描述,但并不因此而限制本發明的保護范圍。以下實施例中所采用的原材料和儀器均為市售。實施例1一種本發明的氮摻雜石墨烯/碳化硅微球納米復合材料,包括碳化硅微球和直立生長于碳化硅微球表面的氮摻雜石墨烯,其中,碳化硅微球為實心結構,碳化硅微球的直徑為0.5μm~1.0μm,氮摻雜石墨烯的長度為0.1μm~0.3μm。一種上述本實施例的氮摻雜石墨烯/碳化硅微球納米復合材料的制備方法,采用如圖1所示的裝置,包括以下步驟:(1)首先將高溫爐升至氮摻雜石墨烯/碳化硅微球的生長溫度,一般為1300℃,然后將液態聚硅烷原料(可市購)通過注射泵以1μL/s的注射速度注射至反應裝置(即高溫爐)中,在氮氣氣氛(也可為氬氣氣氛)中,于1300℃下(1300℃~1400℃均可實施)進行高溫裂解,在石墨片(也可為硅片)表面制備負載石墨烯的碳化硅微球。在高溫爐升溫前,是通過真空泵將爐內空氣抽出,并充入高純氮氣,以制造惰性氣氛。在注射過程結束后,維持高溫爐生長溫度保持0.5小時后,以5℃/min的降溫速率將高溫爐降至室溫。(2)將負載石墨烯的碳化硅微球與質量分數為65%的濃硝酸以10mg∶1mL(10mg~20mg∶1mL~2mL均可實施)的質量體積比混合進行活化處理,超聲分散5min后,于室溫下攪拌96小時,然后離心洗滌至中性,得到負載活化石墨烯的碳化硅微球。(3)將負載活化石墨烯的碳化硅微球與水合肼以10mg∶1mL(10mg~20mg∶1mL~2mL均可實施)的質量體積比混合進行還原處理,超聲分散30min后,先離心洗滌除去水合肼,再于60℃下烘干,然后在氮氣氣氛(也可為氬氣氣氛)中于900℃下保溫2小時(900℃~950℃下保溫2h~3h均可實施),得到氮摻雜石墨烯/碳化硅微球納米復合材料。圖2示出了上述本實施例制得的氮摻雜石墨烯/碳化硅微球納米復合材料的X光電子能譜,圖2中所顯示的4處峰位分別代表氧、氮、碳、硅,其含量也可由X光電子能譜給出,分別為原子百分比10.99%、3.05%、83.12%、2.84%。圖3和圖4為氮摻雜石墨烯/碳化硅微球納米復合材料的掃描電子顯微鏡圖,圖中可清楚地看到,碳化硅微球表面覆蓋生長了直立生長的氮摻雜石墨烯。圖5為氮摻雜石墨烯/碳化硅微球納米復合材料的透射電子顯微鏡圖,由圖5可知,該氮摻雜石墨烯/碳化硅微球表面的氮摻雜石墨烯的長度為0.1μm~0.3μm,碳化硅微球為實心結構,直徑為0.5μm~1.0μm。實施例2一種本發明的氮摻雜石墨烯/碳化硅微球納米復合材料,包括碳化硅微球和直立生長于碳化硅微球表面的氮摻雜石墨烯,其中,碳化硅微球為實心結構,直徑為0.5μm~1.0μm;氮摻雜石墨烯的長度為0.1μm~0.3μm。一種上述本實施例的氮摻雜石墨烯/碳化硅微球納米復合材料的制備方法,包括以下步驟:將液態聚硅烷與乙腈的混合物置于氮氣氣氛(也可為氬氣氣氛)中于1300℃下進行高溫裂解,液態聚硅烷與乙腈的質量比為100∶10,在石墨片(也可為硅片)表面制備得到氮摻雜石墨烯/碳化硅微球納米復合材料。實施例3一種利用實施例1制備的氮摻雜石墨烯/碳化硅微球納米復合材料作為選擇性氧化催化劑在催化反應中的應用,本實施例中的原料為苯甲醇,即苯甲醇的選擇性催化氧化,包括以下步驟:(1)催化劑的磁化:往20mg氮摻雜石墨烯/碳化硅微球納米復合材料中加入1mLFeCl3和FeCl2的混合溶液和10μL氨水(10μL~100μL均可實施,pH達到10左右),混合溶液的摩爾濃度為10mmol/L,FeCl3和FeCl2的摩爾比為1∶1,經超聲分散30min后,用磁鐵分離催化劑和溶液,得到磁化的氮摻雜石墨烯/碳化硅微球納米復合材料。(2)將去離子水、叔丁基過氧化氫和苯甲醇依次加入到磁化的氮摻雜石墨烯/碳化硅微球納米復合材料中,去離子水、叔丁基過氧化氫、苯甲醇和磁化的氮摻雜石墨烯/碳化硅微球納米復合材料的用量比值為3mL∶166μL∶52μL∶20mg,磁化后氮摻雜石墨烯/碳化硅微球納米復合材料重量變化不計,密封后在85℃下保溫反應24小時(即完成選擇性氧化催化反應),完成處理。催化效率檢測利用外加磁場進行催化劑和反應溶液的分離。在溶液中加入0.5mL二氯甲烷萃取產物,然后加入66μL正十二烷作為內標。利用GC-MC(氣質聯用分析儀,型號為:Agilent7890A/5975C,美國安捷倫公司,下同)檢測產物。得到產物峰面積和內標峰面積的比值后(0.001509),與原料(52μL苯甲醇)和內標(66μL正十二烷)經0.5mL二氯甲烷萃取檢測后的比值(1.384758)按照轉化率公式進行計算。轉化率=[1-(產物峰面積/內標峰面積)÷(原料峰面積/內標峰面積)]×100%得到轉化率為99.89%。將分離得到的催化劑洗滌后,加入同樣的原料進行催化反應,重復上述操作5次,得到的轉化率不低于90%,說明本發明的氮摻雜石墨烯/碳化硅微球納米復合材料具有高催化活性、可多次循環使用等優點。實施例4一種利用實施例2制備的氮摻雜石墨烯/碳化硅微球納米復合材料作為選擇性氧化催化劑在催化反應中的應用,本實施例中的原料為乙基苯,即乙基苯的選擇性催化氧化,包括以下步驟:(1)催化劑的磁化:往20mg氮摻雜石墨烯/碳化硅微球納米復合材料中加入1mLFeCl3和FeCl2的混合溶液和10μL~100μL氨水(pH達到10左右),混合溶液的摩爾濃度為10mmol/L,FeCl3和FeCl2的摩爾比為1∶1,經超聲分散30min后,用磁鐵分離催化劑和溶液,得到磁化的氮摻雜石墨烯/碳化硅微球納米復合材料。(2)將去離子水、叔丁基過氧化氫和乙基苯依次加入到磁化的氮摻雜石墨烯/碳化硅微球納米復合材料中,去離子水、叔丁基過氧化氫、乙基苯和磁化氮摻雜石墨烯/碳化硅微球納米復合材料的用量比值為3mL∶166μL∶35μL∶20mg,磁化后氮摻雜石墨烯/碳化硅微球納米復合材料重量變化不計,密封后在85℃保溫反應24小時,完成處理。催化效率檢測將催化后溶液和催化劑用磁鐵分離。在溶液中加入0.5mL二氯甲烷萃取產物,然后加入66μL正十二烷作為內標。利用GC-MC(氣質聯用分析儀)檢測產物。得到產物峰面積和內標峰面積的比值后(0.010874),與原料(35μL乙基苯)和內標(66μL正十二烷)經0.5mL二氯甲烷萃取檢測后的比值(1.06943)按照轉化率公式進行計算。轉化率=[1-(產物峰面積/內標峰面積)÷(原料峰面積/內標峰面積)]×100%得到轉化率為98.98%。將分離得到的催化劑洗滌后,加入同樣的原料進行催化反應,重復上述操作5次,得到的轉化率不低于90%,說明本發明的磁化的氮摻雜石墨烯/碳化硅微球納米復合材料具有高催化活性、可多次循環使用等優點。以上所述僅是本發明的優選實施方式,本發明的保護范圍并不僅局限于上述實施例。凡屬于本發明思路下的技術方案均屬于本發明的保護范圍。應該指出,對于本技術領域的普通技術人員來說,在不脫離本發明原理的前提下的改進和潤飾,這些改進和潤飾也應視為本發明的保護范圍。
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1