二芳基乙烯衍生物與鈣鈦礦材料的復合材料及其制備方法與應用與流程
本發明屬于發光材料技術領域,尤其涉及二芳基乙烯衍生物與鈣鈦礦材料的復合材料及其制備方法與應用。
背景技術:
以金屬鹵化物鈣鈦礦材料作為新型的半導體光電材料引起了學術界和產業界極大地興趣。有機-無機雜化鈣鈦礦材料從分子尺度上結合了有機材料和無機材料的優點,不僅具有無機組分的機械性能和電磁特性,有機組分的已加工成膜等優點,其獨特的無機層和有機層的交替堆積形成的量子阱效應使其在量子約束效應和介電約束效應的雙重作用下,具有很獨特的光電性能。
鈣鈦礦材料不僅在光伏領域具有很大的應用前景,因其半峰寬窄,發光色純度高,適合電子注入的特點,并且可以通過對有機組分和無機組分進行調控從而實現發光特性的控制,在照明,顯示,激光,光探測器等領域具有獨特的應用價值。在有機-無機雜化鈣鈦礦發光領域中,人們采用各種方法來提高有機-無機雜化鈣鈦礦的發光效率。雖然鈣鈦礦材料的熒光量子產率得到了提高,但是只是應用在照明顯示等領域。如何實現對鈣鈦礦材料熒光的調控,增加其用途,未見有報道。
技術實現要素:
本發明的目的是提供二芳基乙烯衍生物與鈣鈦礦材料的復合材料及其制備方法與應用,當用外源交替刺激該復合材料時,可以有效的實現對鈣鈦礦材料熒光的可逆調控,該復合材料在光致熒光分子開關、細胞成像、圖像識別和防偽材料中有大的應用前景。
本發明提供的二芳基乙烯衍生物與鈣鈦礦材料的復合材料,它由二芳基乙烯衍生物和鈣鈦礦材料組成;
所述二芳基乙烯衍生物(分子式為c17h12s2f6r)的結構式如式ⅰ所示:
式ⅰ中,r為甲基、乙基、丙基、羥基、氨基、羧基、甲氧基和醚氧鏈中的任一種;
所述鈣鈦礦材料的分子式為r1nh3ax3、csax3和(r2nh3)2ax4中的任一種,其中,r1為甲基或乙基,r2為碳原子數大于等于4的烷烴有機基團,a為金屬ge、sn、pb、cu、mn、sb和bi中的任一種;x為cl、br和i中的至少一種。
在本發明的具體實施例中,所述二芳基乙烯衍生物的結構式如式ⅰ-a所示:
在本發明的具體實施例中,所述鈣鈦礦材料的分子式可為cspbbr3或ch3nh3pbbr3。
上述的復合材料中,所述二芳基乙烯衍生物和所述鈣鈦礦材料的摩爾比可為(0.1~3):1,具體可為1:1。
上述的復合材料中,所述復合材料可以復合膜的形式存在。
上述的復合材料中,所述鈣鈦礦材料(結晶顆粒)的尺寸大小可為5nm~500nm,具體可為20~40nm。
本發明進一步提供了上述二芳基乙烯衍生物與鈣鈦礦材料的復合材料的制備方法,它包括如下步驟:
(1)制備二芳基乙烯衍生物溶液;
(2)制備鈣鈦礦材料前驅液;
(3)將所述二芳基乙烯衍生物溶液和所述鈣鈦礦材料前驅液混合,除去溶劑,即可得到所述復合材料。
上述的制備方法,步驟(1)中,所述二芳基乙烯衍生物溶液中二芳基乙烯衍生物的濃度可為1mg/ml~100mg/ml,具體可為10mg/ml;溶劑可為n’n-二甲基甲酰胺(dmf)、n-甲基吡咯烷酮(nmp)、γ-丁內酯(γ-bl)、二甲亞砜(dmso)和二甲基乙酰胺(dmac)中的至少一種。
上述的制備方法,步驟(2)中,所述鈣鈦礦材料前驅液的濃度可為0.1%~30%,具體可為1.0%;溶劑可為n’n-二甲基甲酰胺(dmf)、n-甲基吡咯烷酮(nmp)、γ-丁內酯(γ-bl)、二甲亞砜(dmso)和二甲基乙酰胺(dmac)中的至少一種。
所述鈣鈦礦前驅液可按照常規的方法制備得到,具體可通過如下步驟制備得到:
所述分子式為r1nh3ax3的鈣鈦礦材料前驅液的制備方法如下:將鹵化物鹽ax和有機銨鹽r1nh3x分散在溶劑中,即可得到所述分子式為r1nh3ax3的鈣鈦礦材料前驅液;其中,所述鹵化物鹽ax和有機銨鹽r1nh3的摩爾比可為1:(0.5~4),具體可為1:3;所述溶劑可為n’n-二甲基甲酰胺(dmf)、n-甲基吡咯烷酮(nmp)、γ-丁內酯(γ-bl)、二甲亞砜(dmso)和二甲基乙酰胺(dmac)中的至少一種;
所述分子式為csax3的鈣鈦礦材料前驅液的制備方法如下:將鹵化物鹽ax和金屬鹽csx分散在溶劑中,即可得到所述分子式為csax3的鈣鈦礦材料前驅液;其中,所述鹵化物鹽ax和金屬鹽csx的摩爾比可為1:(0.5~4),具體可為1:1;所述溶劑可為n’n-二甲基甲酰胺(dmf)、n-甲基吡咯烷酮(nmp)、γ-丁內酯(γ-bl)、二甲亞砜(dmso)和二甲基乙酰胺(dmac)中的至少一種;
所述分子式為(r2nh3)2ax4的鈣鈦礦材料前驅液的制備方法如下:將鹵化物鹽ax和有機銨鹽r2nh3分散在溶劑中,即可得到所述分子式為(r2nh3)2ax4的鈣鈦礦材料前驅液;其中,所述鹵化物鹽ax和有機銨鹽r1nh3的摩爾比可為1:(0.5~4);所述溶劑可為n’n-二甲基甲酰胺(dmf)、n-甲基吡咯烷酮(nmp)、γ-丁內酯(γ-bl)、二甲亞砜(dmso)和二甲基乙酰胺(dmac)中的至少一種;
r1為甲基或乙基,r2為碳原子數大于等于4的烷烴有機基團,a為金屬ge、sn、pb、cu、mn、sb和bi中的任一種;x為cl、br和i中的至少一種。
上述的制備方法,步驟(3)中,所述方法還包括如下步驟:將所述混合后得到的混合液轉移到基底上制成復合膜。
所述基底可為剛性基底或柔性基底,具體可為聚合物基底;所述聚合物基底可為聚苯乙烯(ps)、聚碳酸酯(pc)、醋酸纖維(ca)、丙烯腈-丁二烯-苯乙烯(abs)、聚氯乙烯(pvc)、聚甲基丙烯酸甲酯(pmma)、聚乙烯(pe)、聚丙烯(pp)、聚氨酯(pu)中的一種或多種。
所述轉移的方法可為旋涂法、噴涂法、刮涂法、絲網印刷法和浸漬提拉法中的任一種。
所述制成復合膜步驟中通過如下方法除去溶劑:將轉移有混合液的基底在30~100℃下加熱0.5~12h,即可得到所述復合膜;具體可在在50~70℃、50℃或70℃下加熱2h。
本發明進一步提供了一種上述的二芳基乙烯衍生物與鈣鈦礦材料的復合材料在下述1)-5)中的至少一種中的應用:
1)制備具有熒光猝滅和恢復(熒光可逆調控)功能的產品;
2)制備熒光分子開關;
3)細胞成像;
4)圖像識別;
5)制備防偽材料。
上述的應用中,對所述復合材料進行外源刺激,即可實現所述復合材料的熒光的猝滅和恢復;所述外源可為光、電、磁、熱和酸堿度中的任一種。具體地,對所述復合材料進行紫外光的刺激,所述復合材料發出的熒光隨著時間的延長而逐漸猝滅;對熒光猝滅后的復合材料進行可見光的刺激,所述猝滅后的熒光隨著時間的延長而逐漸恢復。所述紫外光的波長具體可為254nm。所述可見光的波長具體可為450~650nm。
本發明具有如下有益效果:
本發明復合材料中的鈣鈦礦材料具有良好的熒光發射強度,合成方法簡單普適,較適合工業應用;二芳基乙烯衍生物分子合成路線成熟,產率高,易于放大生產,分子結構容易受光控制。通過紫外光和可見光的交替照射,能夠有效控制二芳基乙烯衍生物分子結構的開環和關環過程,從而實現能量的傳遞,進一步有效控制復合材料中熒光的淬滅和恢復的調控,在光致熒光分子開關、細胞成像、圖像識別和防偽材料中有大的應用前景。
附圖說明
圖1為實施例1中鈣鈦礦材料ch3nh3pbbr3/聚苯乙烯的熒光光譜。
圖2為實施例1中二芳基乙烯衍生物分子2的合成路線。
圖3為實施例1中二芳基乙烯衍生物分子2受紫外或可見光照射,分子發生開環(of-2)和關環(cf-2)結構示意圖。
圖4為實施例1中二芳基乙烯衍生物分子2隨著uv光照射時間的增加,聚苯乙烯基底上二芳基乙烯衍生物2吸收強度的變化圖。
圖5為實施例1中二芳基乙烯衍生物分子2/ch3nh3pbbr3/聚苯乙烯復合膜在254nm光照射下的熒光強度變化圖。
圖6為實施例1中二芳基乙烯衍生物分子2/ch3nh3pbbr3/聚苯乙烯復合膜在450-650nm光照射下的熒光強度變化圖。
圖7為實施例2中鈣鈦礦材料cspbbr3/聚苯乙烯的熒光光譜圖。
圖8為實施例2中二芳基乙烯衍生物分子2/cspbbr3/聚苯乙烯復合膜在254nm光照射下的熒光強度變化圖。
圖9為實施例2中二芳基乙烯衍生物分子2/cspbbr3/聚苯乙烯復合膜在450-650nm光照射下的熒光強度變化圖。
具體實施方式
下述實施例中所使用的實驗方法如無特殊說明,均為常規方法。
下述實施例中所用的材料、試劑等,如無特殊說明,均可從商業途徑得到。
實施例1、制備二芳基乙烯衍生物與鈣鈦礦材料的復合材料
按照如下步驟制備二芳基乙烯衍生物與鈣鈦礦材料的復合材料:
(1)鈣鈦礦材料的制備及熒光光譜分析
將0.11g溴化鉛(pbbr2)與0.10g溴甲胺(ch3nh3br)固體加入樣品瓶中,摩爾比為1:3,然后加入一定2ml的dmf溶劑,超聲后使固體完全溶解,形成透明溶液。然后取0.5ml上述溶液,加入5mldmf溶劑稀釋得到濃度為1.0%的稀前驅溶液,攪拌備用。采用旋涂的方法,將上述稀釋的溶液在2000rmp的轉速下旋涂到1.5*1.5cm大小的聚合物基底上,然后在70℃下加熱2h,去除有機溶劑。在uv-燈下,復合膜為綠光。所使用的聚合物為聚苯乙烯。用熒光光譜儀測試該復合膜的發光在綠光區,發光位置在530nm。圖1為鈣鈦礦材料ch3nh3pbbr3/聚苯乙烯的熒光光譜。
(2)二芳基乙烯衍生物的制備及紫外可見吸收光譜分析
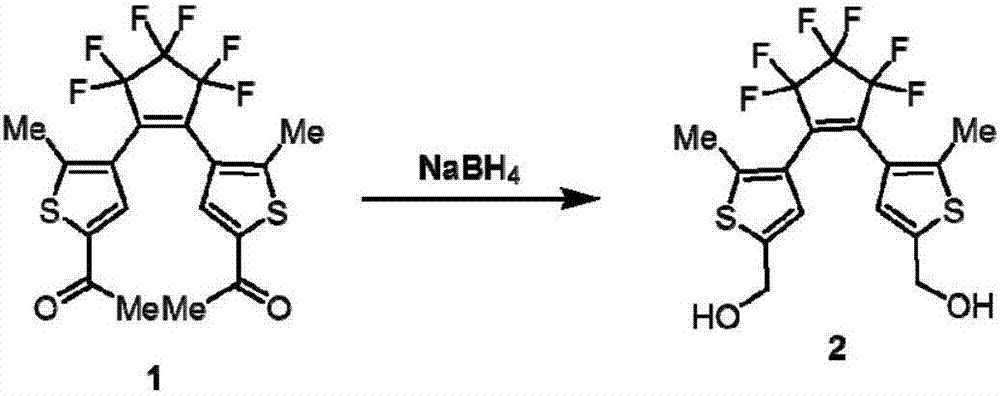
二芳基乙烯衍生物分子2的合成路線如圖2所示,具體步驟如下:向20ml冷甲醇中加入0.24mmol化合物1和0.48mmolnabh4,混合攪拌2h后,加入冷水停止反應。用二氯甲烷萃取產物后,na2so4干燥。采用柱層析提純產物,展開劑為二氯甲烷和甲醇,體積比10:1。其中,化合物1的具體合成路線見文獻j.org.chem.2001,66,3913-3923。
取10mg上述制備得到的二芳基乙烯衍生物分子2,溶解在1mldmf溶劑中,超聲溶解后形成透明溶液,然后將此溶液旋涂到聚合物基底上,50℃加熱2h,去除有機溶劑,測其紫外可見吸收。在400-600nm之間沒有任何吸收。用254nm的光照射二芳基乙烯衍生物分子2/ps,二芳基乙烯衍生物分子2發上關環反應(圖3),測其紫外吸收發現在400-600nm之間逐漸有吸收,且隨著照射時間的增加,吸收強度逐漸增加。圖4為二芳基乙烯衍生物分子2的紫外可見吸收光譜。
(3)復合材料的制備及熒光調控分析
將(2)中制備得到的二芳基乙烯衍生物分子2的dmf溶液和實施例1中濃度為1.0%的ch3nh3pbbr3前驅液以摩爾比為1:1混合。將混合溶液超聲,使其混合均勻。然后將混合溶液采用旋涂的辦法涂布在聚苯乙烯基底上,在50℃加熱2h,去除有機溶劑,得到二芳基乙烯衍生物分子2和ch3nh3pbbr3的復合膜,其中,ch3nh3pbbr3晶體顆粒的尺寸大小約為20nm~40nm,在uv燈下觀察此復合膜為綠色。
測復合膜的熒光光譜,與實施例1中光譜位置一致。采用254nm的紫外光照射復合膜,采集熒光強度隨時間的變化,觀察熒光衰減現象(圖5)。或者采用450-650nm的氙燈照射衰減后的復合膜,測其熒光強度隨照射時間的變化(圖6)。
實施例2、制備二芳基乙烯衍生物與鈣鈦礦材料的復合材料
按照如下步驟制備二芳基乙烯衍生物與鈣鈦礦材料的復合材料:
(1)鈣鈦礦材料的制備及熒光光譜分析
將0.0367g溴化鉛和0.0212g溴化銫放到樣品瓶中混合,控制摩爾比為1:1,然后加入5mldmf溶劑,然后將溶液在80℃下加熱2h,使固體溶解,然后冷卻至室溫備用,制得濃度為~1.0%的鈣鈦礦材料前驅液。采用旋涂的方法,將上述冷卻的溶液在3000rpm轉速下旋涂到1.5*1.5cm大小的聚苯乙烯基底上,然后放到70℃加熱板上加熱2小時,去除有機溶劑。uv燈下復合膜發綠光,用熒光光譜檢測復合膜的發光在綠光區,光譜位置在523nm。圖7為鈣鈦礦cspbbr3/聚苯乙烯的熒光光譜。
(2)二芳基乙烯衍生物的制備及紫外可見吸收光譜分析
同實施例1。
(3)復合材料的制備及熒光調控分析
將(2)中制備得到的二芳基乙烯衍生物分子2的dmf溶液和實施例2中的cspbbr3前驅液以摩爾比為1:1混合。將混合溶液超聲,使其混合均勻。然后將混合溶液采用旋涂的辦法涂布在聚苯乙烯基底上,在70℃加熱2h,去除有機溶劑,得到二芳基乙烯衍生物分子2和cspbbr3的復合膜,其中,cspbbr3晶體顆粒的尺寸大小約為20nm~40nm,在uv燈下觀察此復合膜為綠色。測復合膜的熒光光譜,與實施例2中光譜位置一致。采用254nm的紫外光照射復合膜,采集熒光強度隨時間的變化,觀察熒光衰減現象(圖8)。采用450-650nm的氙燈照射衰減后的復合膜,測其熒光強度隨照射時間的變化(圖9)。
- 還沒有人留言評論。精彩留言會獲得點贊!