一種鑭摻雜碳點的制備方法及其產品、應用與流程
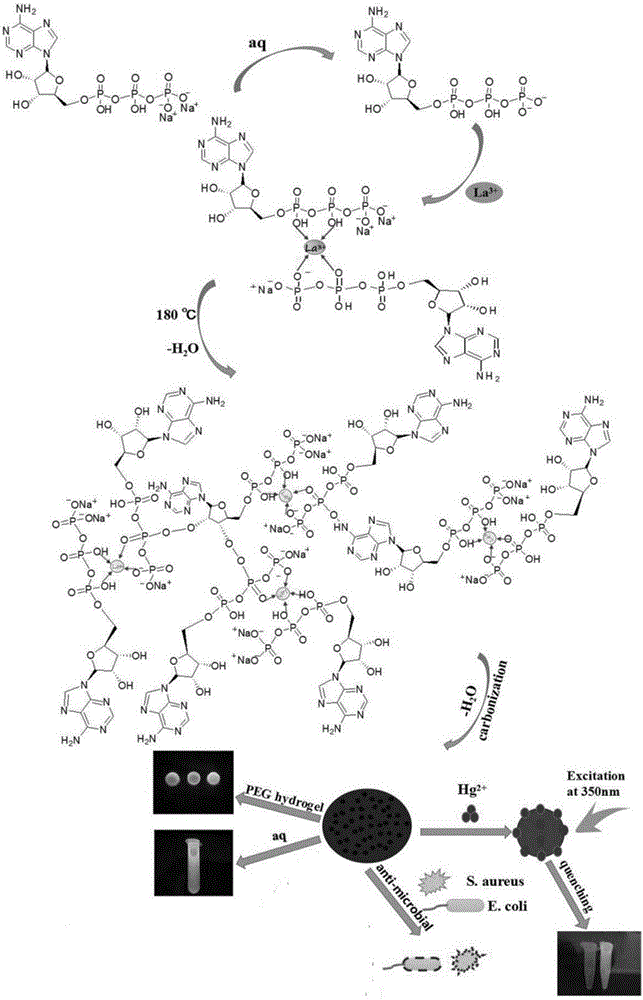
本發明屬于功能納米材料技術領域,具體涉及一種鑭摻雜碳點的制備方法及其產品、應用。
背景技術:
功能納米材料所具備的各種效應,并且獨特的性質使其在生物醫學和農業領域中具有很好的應用前景。其中使用最多的熒光納米粒子就是量子點。量子點是近年來發展起來的一類半導體納米粒子,是半導體介于分子與晶體之間的一種過渡態,具有獨特的光學性質和電學性質。但是量子點的毒性和潛在的環境危害性限制了它的應用。因此尋找適合于生物標記、藥物輸送以及重金屬檢測等生物醫學以及農業領域中所需的納米材料具有重要意義。碳點的出現是納米材料領域的一個新的突破,除了具有傳統半導體量子點所擁有的優良光學性能如熒光穩定性、激發波長依賴性和尺寸小等優點外,還具有傳統半導體量子點無法比擬的高生物相容性、低細胞毒性、無光閃爍、低制備成本及制作工藝相對簡單等優點,在生物醫學和農業領域具有得天獨厚的優勢,因而越來越多地受到科學家的關注。
碳作為一切生物有機體的骨架元素,相對于其他元素構成的熒光納米材料,碳點具有良好的生物相容性,可以通過細胞內吞,進入細胞內部而不影響細胞核,還可以與DNA生物大分子相互作用,從而可以進行DNA的識別與檢測。碳點作為一種新型的金屬離子熒光探針,在溶液中易被電子受體高效猝滅,據此能夠有效地檢測溶液中金屬離子,并在一定范圍內確定金屬離子的濃度,然后進行痕量分析,這在農業重金屬檢測上也有很廣泛的應用。
三磷酸腺苷,也稱腺苷-5’-三磷酸(Adenosine-5’-triphosphate)是一類重要的核苷酸,其基本結構由腺苷和連續三個磷酸基組成,是體內組織細胞一切生命活動所需能量的直接來源,被譽為細胞內的“能量貨幣”。通過高能磷酸鍵的生成和水解,三磷酸腺苷儲存和傳遞著大量化學能,并參與蛋白質、脂肪、糖和核酸的合成,調節細胞的生命過程。因此,三磷酸腺苷及其衍生物被認為可以促使機體各種細胞的修復和再生,增強細胞代謝活性。
技術實現要素:
本部分的目的在于概述本發明的實施例的一些方面以及簡要介紹一些較 佳實施例。在本部分以及本申請的說明書摘要和發明名稱中可能會做些簡化或省略以避免使本部分、說明書摘要和發明名稱的目的模糊,而這種簡化或省略不能用于限制本發明的范圍。
鑒于上述和/或現有鑭摻雜碳點的制備方法的技術空白,提出了本發明。
因此,本發明其中的一個目的是解決現有技術中的不足,提供一種可以特異性檢測Hg2+的存在的鑭摻雜碳點的制備方法。
為解決上述技術問題,本發明提供了如下技術方案:一種鑭摻雜碳點的制備方法,將LaCl3·2H2O和三磷酸腺苷二鈉加入到水中,在15~25℃攪拌反應20~40min,然后在170~190℃的條件下反應6~10h,待其冷卻后過濾,將濾液透析,然后對透析液進行冷凍干燥后得到產品。
作為本發明所述鑭摻雜碳點的制備方法的一種優選方案,其中:所述LaCl3·2H2O和三磷酸腺苷二鈉的質量比為0.5~2:1。
作為本發明所述鑭摻雜碳點的制備方法的一種優選方案,其中:所述水的質量為底物質量的20~40倍。
作為本發明所述鑭摻雜碳點的制備方法的一種優選方案,其中:所述攪拌反應,其攪拌速度為400~600r/min。
作為本發明所述鑭摻雜碳點的制備方法的一種優選方案,其中:所述冷凍干燥,其是在-40~50℃下冷凍干燥20~28h。
作為本發明所述鑭摻雜碳點的制備方法的一種優選方案,其中:所述待其冷卻,其是待其溫度冷卻到20~25℃。
作為本發明所述鑭摻雜碳點的制備方法的一種優選方案,其中:所述過濾,其是用0.22μm的濾膜過濾產物。
作為本發明所述鑭摻雜碳點的制備方法的一種優選方案,其中:所述透析,其是將濾液置于透析袋中透析20~28h。
本發明的另外一個目的是解決現有技術中的不足,提供一種具有良好的生物相容性的鑭摻雜碳點的產品。
為解決上述技術問題,本發明提供了如下技術方案:鑭摻雜碳點組分以質量百分比計,包括碳68~70%、氧10~13%、氮15~17%、磷1.5~3%以及鑭2~2.5%,碳點的直徑尺寸為2.3~2.7nm。
本發明還有一個目的是提供一種鑭摻雜碳點應用于PVA膜方面的方法。 為解決上述技術問題,本發明提供了如下技術方案:將鑭摻雜碳點超聲處理1~3h,稱取PVA顆粒于水中,95~100℃至其完全溶解,得到澄清透明的溶液,將鑭摻雜碳點溶液與PVA水溶液充分攪拌混合,置于室溫環境下干燥成膜,得到PVA/鑭摻雜碳點復合材料。
本發明所具有的有益效果:
(1)本發明制備的碳點不僅可以特異性檢測Hg2+的存在,并且隨著Hg2+的濃度增加,碳點的熒光強度不斷地降低,可應用于重金屬檢測。
(2)該碳點可以有效的抑制細菌的生長,并且通過細胞毒性試驗證明其具有良好的生物相容性,能夠很好地應用于醫用傷口敷料以及食品包裝中。
(3)本發明制備碳點的產率很高,最高可至57%。
(4)本發明制備的碳點熒光具有激發波長依賴性,這一系列的結果為碳點藥物運輸、生物成像、發光材料的制備等方面的應用提供了新的平臺。
(5)本發明制備的碳點在水溶液中具有良好的分散性,其生物相容性優越,在生物醫藥和農業領域均有著廣闊的應用前景。
附圖說明
為了更清楚地說明本發明實施例的技術方案,下面將對實施例描述中所需要使用的附圖作簡單地介紹,顯而易見地,下面描述中的附圖僅僅是本發明的一些實施例,對于本領域普通技術人員來講,在不付出創造性勞動性的前提下,還可以根據這些附圖獲得其它的附圖。其中:
圖1是本發明制備的La-CDs的反應過程示意圖。
圖2是本發明制備的La-CDs的透視電鏡圖(插圖為碳點的粒徑分布圖)。可以很好的看出,該碳點表現為相對均一的球形顆粒,具有很好的分散性,且碳點的粒徑呈正態分布,納米碳點的直徑尺寸大約為2.5nm。
圖3A和3B是本發明制備的碳點的激發波長依賴發射光譜和相應的標準化發射光譜圖(實施例6)。La-CDs的熒光強度隨著激發波長的增加呈先上升后下降的趨勢,且最大的激發波長為350nm,最大發射波長為470nm。碳點的發射波長隨著激發波長的增加而逐漸紅移,呈現多元激發、多元發射的光譜特性。
圖4A和4B是實施例7中不同金屬離子對碳點的熒光強度的影響和不同濃度的Hg2+對碳點的熒光相應圖。在Hg2+,Ag+,Ba2+,Ca2+,Cd2+,Co2+,Cu2+,Fe3+,Mg2+,Mn2+,Ni2+,Ca2+,Na+,Pb2+和Zn2+的存在下,只有Hg2+對碳點的熒光有淬滅作用,并且隨著Hg2+的濃度增加,碳點的熒光強度不斷下降。表明了所制備的碳點對Hg2+有著特異的識別。
圖5A和5B是本發明制備的碳點表面粘附大腸桿菌以及金色葡萄球菌的SEM圖(實施例8)。我們可以很明顯的看出單純的PVA表面有著大量的正常生長的大腸桿菌以及金色葡萄球菌,但是含有La-CDs的PVA膜表面則大腸桿菌以及金色葡萄球菌粘附含量較少,均呈現死亡狀態,且大腸桿菌已經金色葡萄球菌的形態發生了一些變化,結構逐漸塌陷。
圖6為實施例1~4制得的La-CDs的熒光強度對比圖。
圖7為實施例9中相對增殖率的柱狀圖。
具體實施方式
為使本發明的上述目的、特征和優點能夠更加明顯易懂,下面結合具體實施例對本發明的具體實施方式做詳細的說明。
在下面的描述中闡述了很多具體細節以便于充分理解本發明,但是本發明還可以采用其他不同于在此描述的其它方式來實施,本領域技術人員可以在不違背本發明內涵的情況下做類似推廣,因此本發明不受下面公開的具體實施例的限制。
其次,此處所稱的“一個實施例”或“實施例”是指可包含于本發明至少一個實現方式中的特定特征、結構或特性。在本說明書中不同地方出現的“在一個實施例中”并非均指同一個實施例,也不是單獨的或選擇性的與其他實施例互相排斥的實施例。
實施例1
摻雜鑭元素的碳點采用如下方法制備:
第一步:將0.5g LaCl3·2H2O和1g三磷酸腺苷二鈉(ATP)分別加入30g去離子水中溶解;
第二步:將LaCl3·2H2O和三磷酸腺苷二鈉(ATP)的水溶液混合,在20℃下,以600r/min攪拌30min。
第三步:將溶液轉移到反應釜中,在180℃的條件下反應8h。
第四步:待到反應釜冷卻到20℃時,用0.22μm的濾膜過濾產物,再將濾液置于透析袋中透析24h,然后將得到的透析液在-50℃下冷凍干燥24h得到固 態的La-CDs(lanthanum-carbon dots)。
用X射線光電子能譜分析(XPS)直接測試所制得的La-CDs,其檢測結果為,該La-CDs包括碳68.85%、氧11.85%、氮15.32%、磷1.58%以及鑭2.40%。
通過得到的產物質量比上反應物的質量,得碳點產率為55.65%。
實施例2
摻雜鑭元素的碳點采用如下方法制備:
第一步:將1.0g LaCl3·2H2O和1g三磷酸腺苷二鈉(ATP)分別加入40g去離子水中溶解;
第二步:將LaCl3·2H2O和三磷酸腺苷二鈉(ATP)的水溶液混合,在20℃下,以500r/min攪拌25min。
第三步:將溶液轉移到反應釜中,在190℃的條件下反應8h。
第四步:待到反應釜冷卻到25℃時,用0.22μm的濾膜過濾產物,再將濾液置于透析袋中透析24h,然后將得到的透析液在-55℃下冷凍干燥24h得到固態的La-CDs。
用X射線光電子能譜分析(XPS)直接測試所制得的La-CDs,其檢測結果為,該La-CDs包括68.22%、氧10.45%、氮16.46%、磷2.85%以及鑭2.02%。
通過得到的產物質量比上反應物的質量,得碳點產率為54.89%。
實施例3
摻雜鑭元素的碳點采用如下方法制備:
第一步:將1.5g LaCl3·2H2O和1g三磷酸腺苷二鈉(ATP)分別加入50g去離子水中溶解;
第二步:將LaCl3·2H2O和三磷酸腺苷二鈉(ATP)的水溶液混合,在20℃下,以600r/min攪拌40min。
第三步:將溶液轉移到反應釜中,在180℃的條件下反應8h。
第四步:待到反應釜冷卻到20℃時,用0.22μm的濾膜過濾產物,再將濾液置于透析袋中透析24h,然后將得到的透析液-55℃下冷凍干燥28h得到固態的La-CDs。
用X射線光電子能譜分析(XPS)直接測試所制得的La-CDs,其檢測結果為,該La-CDs包括碳69.02%、氧10.43%、氮16.52%、磷1.56%以及鑭2.47%。
通過得到的產物質量比上反應物的質量,得碳點產率為57.03%。
實施例4
摻雜鑭元素的碳點采用如下方法制備:
第一步:將2.0g LaCl3·2H2O和1g三磷酸腺苷二鈉(ATP)分別加入60g去離子水中溶解;
第二步:將LaCl3·2H2O和三磷酸腺苷二鈉(ATP)的水溶液混合,在15℃下,以500r/min攪拌30min。
第三步:將溶液轉移到反應釜中,在170℃的條件下反應9h。
第四步:待到反應釜冷卻到20℃時,用0.22μm的濾膜過濾產物,再將濾液置于透析袋中透析24h,然后將得到的透析液-50℃下冷凍干燥24h得到固態的La-CDs。
用X射線光電子能譜分析(XPS)直接測試所制得的La-CDs,其檢測結果為,該La-CDs包括碳68.02%、氧12.00%、氮16.02%、磷1.70%以及鑭2.26%。
通過得到的產物質量比上反應物的質量,得碳點產率為56.11%。
將實施例1~4制得的La-CDs配制0.5mg/ml的溶液,用熒光分度計測量其熒光強度如圖6所示。其結果為,實施例3中制得的La-CDs熒光強度最高。
實施例5
取適量稀濃度La-CDs水溶液滴于覆蓋碳膜的銅網上,室溫晾干后用透射電鏡(日本Hitachi公司生產的H-7650型)觀察樣品形態,見附圖2。可以看出,該碳點的直徑大約在2.3~2.7nm,且分散均勻,由此可見,該發明制得的La-CDs具有尺寸均一以及很好的分散性優點。
實施例6
用去離子水配制0.5mg/ml合成的碳點溶液,用熒光分度計測量不同激發波長下的熒光發射譜圖。如附圖3所示,在不同激發波長下的熒光發射波發生了轉移。La-CDs的熒光強度隨著激發波長的增加呈先上升后下降的趨勢,且最大的激發波長為350nm,最大發射波長為470nm。碳點的發射波長隨著激發波長的增加而逐漸紅移,呈現多元激發、多元發射的光譜特性。
由此可見,利用本發明提供的制備方法制得的碳點具有激發波長依賴性,可以在不同的激發下呈現出不同的顏色。
實施例7
將合成的碳點溶液配制0.1mM的Hg2+,Ag+,Ba2+,Ca2+,Cd2+,Co2+,Cu2+,Fe3+,Mg2+,Mn2+,Ni2+,Ca2+,Na+,Pb2+和Zn2+溶液,然后用熒光分度計測量其溶液的熒光強度(如圖4A)。
用合成的碳點溶液配制了不同濃度的Hg2+溶液,然后用熒光分度計測量其熒光強度(如圖4B)。我們發現只有汞離子溶液的熒光有大幅度的降低,其他離子溶液熒光強度沒有明顯的變化。并且隨著汞離子的濃度增加,其熒光強度不斷下降。在Hg2+,Ag+,Ba2+,Ca2+,Cd2+,Co2+,Cu2+,Fe3+,Mg2+,Mn2+,Ni2+,Ca2+,Na+,Pb2+和Zn2+的存在下,只有Hg2+對碳點的熒光有淬滅作用,并且隨著Hg2+的濃度增加,碳點的熒光強度不斷下降。表明了所制備的碳點對Hg2+有著特異的識別。
由此可見,利用本發明提供的制備方法制得的碳點可以特異性識別汞離子,并且可以檢測出其濃度。
實施例8
將合成的La-CDs于去離子水中超聲分散2h。稱取適量PVA顆粒于去離子水中,96℃恒溫水浴至其完全溶解,得到澄清透明的溶液。將不同體積的改性氧化石墨烯溶液與PVA水溶液充分攪拌混合,倒入聚四氟乙烯表面皿,置于室溫環境下干燥成膜,得到PVA/鑭摻雜碳點復合材料:PVA/La-CDs。
將PVA、PVA/La-CDs薄膜裁成直徑為8mm的膜片,浸泡于PBS中24h,自然晾干,紫外照射殺菌30min后放入24孔板中,加入2ml液體培養基和20μl活化24h的大腸桿菌(E.coli)和金黃色葡萄球菌(S.aureus)菌液,用塞子封口,置于37℃恒溫振蕩培養24h。然后用大量PBS沖去粘附不牢的細菌,用2.5%的戊二醛溶液(v/v,PBS稀釋)固定膜片上的細菌4h,之后分別用50%、60%、70%、80%、90%和100%梯度濃度的乙醇溶液逐級脫水,每次浸泡30min。最后將膜片置于超凈臺自然干燥,噴金后用日本電子公司生產的JSM-5610LV型的掃描電子顯微鏡觀察材料表面的細菌粘附情況。
如附圖5所示,我們可以很明顯的看出單純的PVA表面有著大量的正常生長的大腸桿菌以及金色葡萄球菌,但是含有La-CDs的PVA膜表面則大腸桿菌以及金色葡萄球菌粘附含量較少,均呈現死亡狀態,且大腸桿菌已經金色葡萄球菌的形態發生了一些變化,結構逐漸模糊。其結果為,PVA/La-CDs薄膜表面的大腸桿菌和金黃色葡萄球菌的數目明顯少于對照組PVA薄膜。
由此可見,含有本發明制得的La-CDs的PVA膜表面對革蘭氏陽性與陰性 菌具有抑制作用。
實施例9
用DMEM配制待測樣品的梯度濃度溶液,依次為500μg/mL、250μg/mL、100μg/mL和50μg/mL。
將培養好的NIH3T3細胞用0.25%的胰蛋白酶進行消化,充分吹打使其形成均勻地單細胞懸液后,以100μL/well的量加入96孔板中。將96孔板置于CO2培養箱中繼續培養至細胞鋪滿孔底80%~90%時,吸出原有培養液,加入等量的不同濃度的樣品溶液,每組接種3~6孔。給藥后將孔板置于CO2培養箱中培養24h,拿出后每孔加入20μL濃度為5mg/mL的MTT溶液,37℃恒溫接觸4h后,小心除去孔內液體,并加入150μL DMSO,室溫下輕微振蕩,使底部晶體充分溶解,用酶標儀在570nm波長下測定每孔的O.D.值。然后利用O.D.值來計算細胞相對增殖率。
如附圖7所示,其結果為,La-CDs沒有明顯的細胞毒性,生物相容性良好,可用作生物醫用材料。
綜上所述,通過本發明所述的制備方法制得的碳點添加到膜中,不僅可以特異性檢測Hg2+的存在,并且隨著Hg2+的濃度增加,碳點的熒光強度不斷地降低;該碳點可以有效的抑制細菌的生長,并且通過細胞毒性試驗證明其具有良好的生物相容性,添加到膜中能夠很好地應用于醫用傷口敷料以及食品包裝中;同時本發明制備的碳點的產率很高,大約在57%;由于該碳點熒光具有激發波長依賴性,這一系列的結果為碳點藥物運輸、生物成像、發光材料的制備等方面的應用提供了新的平臺;最后,本發明制備的碳點在水溶液中具有良好的分散性,生物相容性優越,在生物材料領域有著廣闊的應用前景。
應說明的是,以上實施例僅用以說明本發明的技術方案而非限制,盡管參照較佳實施例對本發明進行了詳細說明,本領域的普通技術人員應當理解,可以對本發明的技術方案進行修改或者等同替換,而不脫離本發明技術方案的精神和范圍,其均應涵蓋在本發明的權利要求范圍當中。
- 還沒有人留言評論。精彩留言會獲得點贊!