一種二價鎳離子激活的近紅外長余輝納米材料及其制備方法和應用與流程
本發明涉及近紅外長余輝材料,特別涉及一種二價鎳離子激活的近紅外長余輝納米材料及其制備方法和應用。
背景技術:
光學成像以光子作為信息源,代表了一個快速延伸的領域并被直接應用于藥理學、分子細胞生物學和診斷學。但是這種技術仍然存在許多局限性,尤其是在體內光照時產生的組織自發熒光和在短波激發光照射下的弱的組織滲透性。為了克服這些困難,科學家研究了一系列無機發光材料,發射光是在近紅外區域(NIR),分子發射近紅外光(650-1350nm),可以用于活體分子目標的探測,因為生物體血液和組織在這個波長范圍內內是相對透明的,從而減少了體內背景干擾造成的難題。而長余輝材料因為在信號采集過程中沒有激發光的干擾,可以極大限度的提高成像精度。但是現在這種近紅外材料的種類還是很少,發光中心也很少。最近發展的近紅外長余輝材料也僅限于高溫固相燒結,高溫燒結將導致長余輝材料顆粒增大,因此要想進一步推進近紅外長余輝材料在生物成像中的應用,非常有必要發展改進的合成技術制備納米近紅外長余輝材料。
技術實現要素:
為了克服現有技術的上述缺點與不足,本發明的目的在于提供一種二價鎳離子激活的近紅外長余輝納米材料,余輝帶寬為1050-1600納米,余輝峰位于1250-1350納米處。
本發明的目的通過以下技術方案實現:
一種二價鎳離子激活的近紅外長余輝納米材料,以ZnGa2O4為基體材料,基體材料中摻雜0.1~5mol%的Ni。
所述的二價鎳離子激活的近紅外長余輝納米材料,包括以下步驟:
(1)以醋酸鋅,硝酸鎵,硝酸鎳為原料,將原料加入水和酒精的混合溶液中,在室溫下攪拌后加入乙酰丙酮,在室溫下攪拌得到混合溶液,其中混合溶液的PH值控制在2-4;
(2)將步驟(1)得到的混合溶液放到80-90℃烘箱干燥24-36h,得到濕凝膠;
(3)將正丁醇和酒精的混合溶液加入到步驟(2)得到的濕凝膠中,放到70-80℃的油浴鍋中反應1-2h,隨后升溫到95-100℃反應1-1.5小時,待燒杯中的液體完全反應后,將溫度提升到120-150℃反應20-30分鐘,干燥后得到干凝膠;
(4)步驟(3)得到的干凝膠經研磨后轉移到坩堝中,在900-1200℃熔爐中燒1-2h,得到粉體材料。
所述的二價鎳離子激活的近紅外長余輝納米材料,還包括以下步驟:
(5)將步驟(4)得到的粉體裝入離心管,通過超聲30min,6000-7000轉離心10-15min,酒精洗3-4次,水洗3-4次,之后靜置至少48小時后以10000轉離心10-15min,不同粒徑的粉體顆粒分離,得到顆粒小于100納米的納米粒子。
步驟(1)所述乙酰丙酮與混合溶液中的金屬離子總量的摩爾比為1:1。
步驟(1)所述水和酒精的混合溶液中,水和酒精的體積比為=1:(1-1.5)。
步驟(3)所述的正丁醇和酒精的混合溶液中,正丁醇百分比為30-40%。
所述二價鎳離子激活的近紅外長余輝納米材料的應用,用于作為生物成像中的標記物。
與現有技術相比,本發明具有以下優點和有益效果:
(1)本發明的近紅外長余輝納米材料,通過摻雜Ni2+離子,實現了近紅外長余輝發光,余輝帶寬為1050-1600納米,余輝峰位于1250-1350納米處。
(2)本發明的近紅外長余輝納米材料的制備方法,制備得到的近紅外長余輝粉體材料,顆粒小于100納米并且單分散性良好,均勻性優異,顆粒經高溫1000度以上高溫燒結后,仍然不會團聚,顆粒穩定性良好,且余輝時間大于350分鐘,能夠很好的應用于生物成像領域。
附圖說明
圖1為本發明的實施例1制備的納米近紅外長余輝材料的熒光發射光譜。
圖2為本發明的實施例1制備的納米近紅外長余輝材料的熒光激發光譜。
圖3為本發明的實施例1制備的納米近紅外長余輝材料的余輝光譜。
圖4為本發明的實施例1制備的納米近紅外長余輝材料的余輝衰減曲線。
圖5為本發明的實施例1制備的納米近紅外長余輝材料的掃描電鏡圖。
圖6為本發明的實施例2制備的納米近紅外長余輝材料的熒光發射光譜。
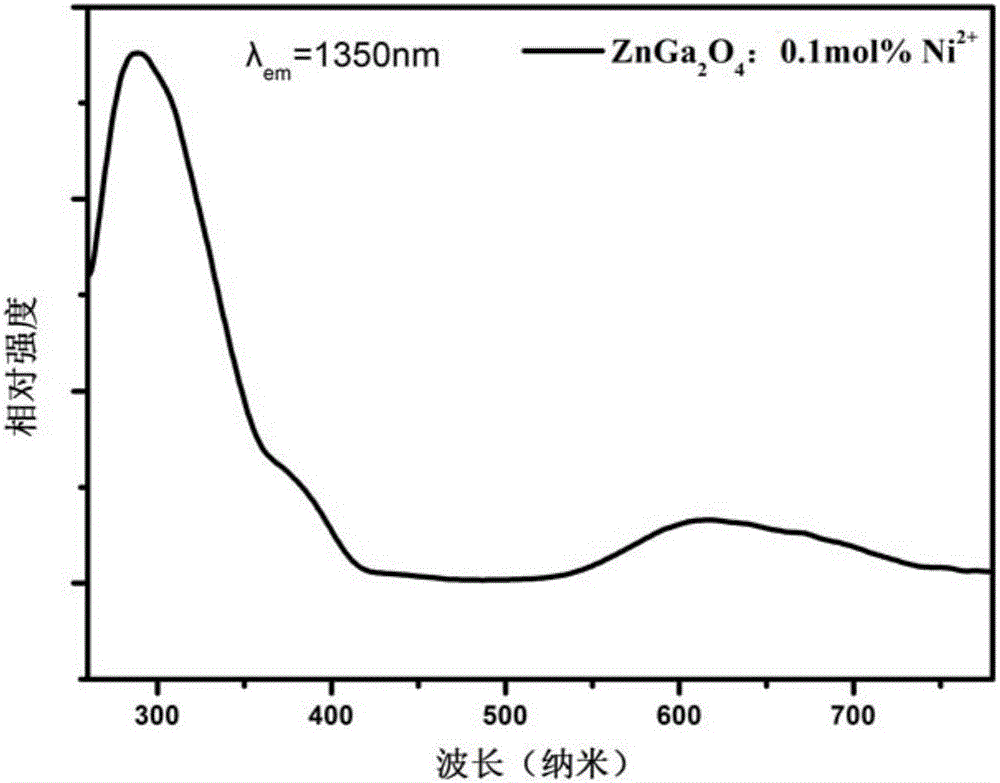
圖7為本發明的實施例2制備的納米近紅外長余輝材料的熒光激發光譜。
圖8為本發明的實施例2制備的納米近紅外長余輝材料的余輝光譜。
圖9為本發明的實施例2制備的納米近紅外長余輝材料的余輝衰減曲線。
圖10為本發明的實施例2制備的納米近紅外長余輝材料的掃描電鏡圖。
具體實施方式
下面結合實施例,對本發明作進一步地詳細說明,但本發明的實施方式不限于此。
實施例1
本實施例的Ni2+離子激活鎵酸鋅納米近紅外長余輝標記物的制備方法:
按照以下成分:基體為ZnGa2O4,Ni2+的摻雜量為0.1mol%;
(1)以醋酸鋅,硝酸鎵,硝酸鎳為原料,稱取原料并加入20g水和酒精的混合溶液中,在室溫下攪拌30分鐘后加入取乙酰丙酮,在室溫下攪拌1小時得到混合溶液,其中混合溶液的PH值控制在2;其中,乙酰丙酮與混合溶液中的金屬離子總量的摩爾比為1:1;所述水和酒精的混合溶液中,水和酒精的體積比為=1:1;
(2)將步驟(1)得到的混合溶液放到80℃烘箱干燥24h,得到濕凝膠;
(3)將20g正丁醇和酒精的混合溶液加入到步驟(2)得到的濕凝膠中,放到70℃的油浴鍋中反應1h,隨后升溫到98℃反應1小時,待燒杯中的液體完全反應后,將溫度提升到150℃反應20分鐘,干燥后得到干凝膠;所述正丁醇和酒精的混合溶液中,正丁醇和酒精的體積比為3:7;
(4)步驟(3)得到的干凝膠經研磨后轉移到坩堝中,在1200℃熔爐中燒2h,得到粉體;
(5)將步驟(4)得到的粉體裝入離心管,通過超聲30min,6000轉離心10min,酒精洗3次,水洗3次,之后靜置48小時后通過10000轉離心10min將大顆粒和小顆粒進行分離,即可得到顆粒小于100納米,單分散良好的納米粒子。
本實施例制備的Ni2+摻雜的鎵酸鋅納米近紅外長余輝納米材料的熒光光譜如圖1所示顯示,在300nm激發下出現了1250nm的發光。圖2是監測1250nm的發光,有3個發光峰,分別位于300nm、380nm和600nm。這3個激發峰都是Ni2+的特征激發峰。圖3顯示了本實施例制備的Ni2+摻雜的鎵酸鋅納米近紅外長余輝納米材料在300nm光下照射10分鐘后,所制備樣品具有1050-1600的寬帶余輝發光,并且余輝峰位于1250nm處。圖4顯示停止激發后監測1250nm的發光,衰減時間長達350分鐘。圖5是本發明制備的Ni2+摻雜的鎵酸鋅納米近紅外長余輝納米材料的掃描電鏡圖,顯示了本發明制備的材料顆粒小于100納米,并且分散性良好,單顆納米粒子的形貌可見,雖然經過了1200攝氏度的高溫燒結,但是依然能夠得到分散性很好的納米粒子,使得這種材料能夠很好的應用在生物成像中。
實施例2
本實施例的Ni2+離子激活鎵酸鋅納米近紅外長余輝標記物的制備方法:
按照以下成分:基體為ZnGa2O4,Ni2+的摻雜量為5mol%;
(1)以醋酸鋅,硝酸鎵,硝酸鎳為原料,稱取原料并加入20g水和酒精的混合溶液中,在室溫下攪拌30分鐘后加入取乙酰丙酮,在室溫下攪拌1小時得到混合溶液,其中混合溶液的PH值控制在4;其中,乙酰丙酮與混合溶液中的金屬離子總量的摩爾比為1:1;所述水和酒精的混合溶液中,水和酒精的體積比為=1:1;
(2)將步驟(1)得到的混合溶液放到80℃烘箱干燥24h,得到濕凝膠;
(3)將20g正丁醇和酒精的混合溶液加入到步驟(2)得到的濕凝膠中,放到70℃的油浴鍋中反應1h,隨后升溫到98℃反應1小時,待燒杯中的液體完全反應后,將溫度提升到150℃反應20分鐘,干燥后得到干凝膠;所述正丁醇和酒精的混合溶液中,正丁醇和酒精的體積比為3:7;
(4)步驟(3)得到的干凝膠經研磨后轉移到坩堝中,在900℃熔爐中燒2h,得到粉體;
(5)將步驟(4)得到的粉體裝入離心管,通過超聲30min,6000轉離心10min,酒精洗3次,水洗3次,之后靜置48小時后通過10000轉離心10min將大顆粒和小顆粒進行分離,即可得到顆粒小于100納米,單分散良好的納米粒子。
本實施例制備的Ni2+摻雜的鎵酸鋅納米近紅外長余輝納米材料的熒光光譜如圖6所示顯示,在300nm激發下出現了1350nm的發光。圖7是監測1250nm的發光,有3個發光峰,分別位于300nm、390nm和620nm。這3個激發峰都是Ni2+的特征激發峰。圖8顯示了本實施例制備的Ni2+摻雜的鎵酸鋅納米近紅外長余輝納米材料在300nm光下照射10分鐘后,所制備樣品具有1050-1600的寬帶余輝發光,并且余輝峰位于1350nm處。圖9顯示停止激發后監測1350nm的發光,衰減時間長達150分鐘。圖10是本發明制備的Ni2+摻雜的鎵酸鋅納米近紅外長余輝納米材料的掃描電鏡圖,顯示了本發明制備的材料顆粒小于100納米,并且分散性良好,單顆納米粒子的形貌可見,雖然經過了900攝氏度的高溫燒結,但是依然能夠得到分散性很好的納米粒子,使得這種材料能夠很好的應用在生物成像中。
上述實施例為本發明較佳的實施方式,但本發明的實施方式并不受所述實施例的限制,其他的任何未背離本發明的精神實質與原理下所作的改變、修飾、替代、組合、簡化,均應為等效的置換方式,都包含在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!