一種黑磷量子點及其制備方法與流程
本發明涉及新型納米材料領域,特別是涉及一種黑磷量子點及其制備方法。
背景技術:
作為磷的一種同素異形體,新型二維層狀材料—黑磷,具有類似但不同于石墨烯片層結構的天然褶皺結構,這種獨特的褶皺結構導致了黑磷擁有許多獨特的物理性質,如黑磷具備石墨烯所沒有的半導體間隙且是直接帶隙,即電子導電能帶(導帶)底部和非導電能帶(價帶)頂部在同一位置,實現從非導電導導電,電子值需要吸收能力,這意味著黑磷可用作可調半導體,與光可以直接耦合,光譜包括了整個可見光到近紅外區域。因此,黑磷在納米光電子學、生物醫學等領域具有廣闊的應用前景,如:光電器件、傳感器、開關等。而黑磷量子點因為其具有超小的尺寸在光學標記、癌癥治療等方面更具有應用前景,因此,黑磷量子點的制備極具現實意義。
目前,通過機械剝離法(如透明膠帶撕分法)、化學氣相沉積法等技術,學者們已經制備出片狀的黑磷材料。但是,機械剝離法制備的片狀黑磷的產率較低,不適合用來生產商業應用性材料,且操作繁瑣、耗時長、得到的材料尺寸較大,多在微米級;而化學氣相沉積法制備的黑磷片的量非常少,且不易重復。因此,急需開發出一種大規模制備小尺寸黑磷量子點的便捷方法。
技術實現要素:
有鑒于此,本發明提供了一種黑磷量子點及其制備方法,該制備方法通過將研磨后的塊體黑磷分散在有機溶劑中,并置于高溫下的密閉容器中加熱一定時間,可以獲得分散性較好、尺寸均一的黑磷量子點,所制得的黑磷量子點的尺寸較小,約在1~5nm,且在紫外可見光區域呈現出明顯的吸收特性。該方法工藝簡單易操作,產率較高、重現性好,并且可實現大規模制備,對環境不產生二次污染問題。
第一方面,本發明提供了一種黑磷量子點的制備方法,包括以下步驟:
(1)在無氧條件下,將粒徑為5~50μm的黑磷分散在第一有機溶劑中,得到第一分散液,所述第一分散液中黑磷的濃度為0.6~1.2mg/mL;
(2)采用所述第一有機溶劑對所述第一分散液稀釋5~10倍,之后加入氫氧化鈉,得到第二分散液,所述第二分散液中,黑磷與氫氧化鈉的質量比為0.006~0.012:1;
(3)將所述第二分散液置于密閉容器中,在溫度為120~160℃下加熱反應6~18h,反應完畢后,將所得反應液冷卻至室溫,離心,收集上清液,得到黑磷量子點。
優選地,所述第二分散液中黑磷的濃度為0.06~0.12mg/mL。
步驟(2)中,第二分散液中加入有氫氧化鈉,一方面氫氧化鈉中的鈉離子和氫氧根離子能夠插層到黑磷片層中,削弱黑磷片層之間的范德華力,提高了剝離效率,另一方面多余的氫氧根離子可以吸附在黑磷表面起到保護黑磷的作用,提高了黑磷的穩定性。因此,本申請中優選使第二分散液中黑磷的質量與所述氫氧化鈉的質量比為0.006~0.012:1。
另外,如果氫氧化鈉濃度太低,則其插層到黑磷片層中的概率將會降低,所以為了達到更好地剝離效果,一般加入的氫氧化鈉會過量一些。但若加入太多NaOH的話,因氫氧化鈉在第二分散液中的溶解度不是很高,加入太多會較浪費物料。
優選地,步驟(2)中,取所述第一分散液2~4mL,再加入18~36mL的所述第一有機溶劑,之后加入200~400mg的氫氧化鈉,得到第二分散液。
本申請中,步驟(2)中,在溫度為120~160℃下加熱6~18h來進行黑磷的剝離,高溫提供的能量能夠削弱黑磷片層之間的范德華力,使其更容易剝離。
優選地,步驟(2)中,所述密閉容器為高壓反應釜(例如聚四氟乙烯反應釜)或三口瓶。
優選地,當所述密閉容器為三口瓶時,還在所述加熱反應的過程中進行攪拌,其中,所述攪拌的速度為3000-4000轉/分。
優選地,所述黑磷量子點的尺寸在1~5nm。進一步優為2-4nm。
優選地,步驟(1)中,所述粒徑為5~50μm的黑磷是通過將黑磷塊體進行研磨得到。
進一步優選地,所述黑磷塊體的研磨是在真空環境下(無氧環境下)進行的。優選在有氮氣的保護的手套箱中進行,并且在研磨過程中加入一些第一有機溶劑。
更優選地,所述黑磷塊體是利用白磷在高壓強和高溫度下轉化而形成。
優選地,所述第一有機溶劑包括N-甲基吡咯烷酮(NMP)、二甲基甲酰胺(DMF)、二甲基亞砜(DMSO)、二甲基咪唑啉(DMEU)、丙酮和無水乙醇中的一種或多種。
更優選地,所述第一有機溶劑為N-甲基吡咯烷酮。
優選地,所述加熱反應的溫度為120~135℃。
進一步優選地,所述加熱反應的溫度為120~130℃。
優選地,所述加熱反應的溫度為135~160℃。
進一步優選地,所述加熱反應的溫度為140~160℃。
優選地,步驟(2)中,所述加熱反應的時間為6~12h。
進一步優選地,所述加熱反應的時間為8~12h。
優選地,所述加熱反應的時間為14~18h。
進一步優選地,所述加熱反應的時間為16~18h。
本發明實施例中,所述加熱反應的溫度為120℃、125℃、130℃、135℃、140℃、150℃、160℃。所述加熱反應的時間可以為6h、8h、10h、12h、14h、16h、18h。
優選地,步驟(3)中,所述離心的轉速為6000~9000轉/分鐘,離心時間為10-30分鐘。優選為15-25分鐘。
優選地,步驟(3)中,所述離心是在手套箱中氮氣保護下進行。
優選地,所述步驟(3)還包括:
在所述收集上清液之后,對所述上清液進行高速離心10-30min,收集固體產物或將所述固體產物分散到第二有機溶劑中,得到黑磷量子點。
優選地,所述高速離心的轉速為10000-12000rpm。
優選地,所述第二有機溶劑包括包括N-甲基吡咯烷酮(NMP)、二甲基甲酰胺(DMF)、二甲基亞砜(DMSO)、二甲基咪唑啉(DMEU)、丙酮和無水乙醇中的一種或多種。
所述第一有機溶劑和所述第二有機溶劑,可以為同種溶劑,也可以為不同種溶劑。
本發明第一方面提供的黑磷量子點的制備方法,以實現對黑磷塊體的剝離,此方法工藝簡單易操作,可以進行批量化剝離,剝離產率較高,相比目前的機械剝離法及化學氣相沉積法等有明顯的優勢,可獲得尺寸可控的單分散性好、尺寸均一的小尺寸黑磷量子點,易實現低成本產業化生產。
基于本發明第一方面的制備方法,可以實現從黑磷固體到黑磷量子點的大規模產業化制備,為黑磷量子點在生物醫學領域、光電子領域的應用奠定基礎。
第二方面,本發明還提供了由上述制備方法制備得到的黑磷量子點。所述黑磷量子點的尺寸在1~5nm。
所制得的黑磷量子點的粒徑分布較窄,尺寸較小,在紫外-可見光區域表現出明顯的吸收特性,可以應用于制備光熱治療藥物、光學標記藥物、傳感器等生物醫學前沿領域。
本發明實施例的優點將會在下面的說明書中部分闡明,一部分根據說明書是顯而易見的,或者可以通過本發明實施例的實施而獲知。
附圖說明
圖1為本發明實施例1制得的黑磷量子點在N-甲基吡咯烷酮溶液中的紫外-可見-近紅外吸收光譜圖;
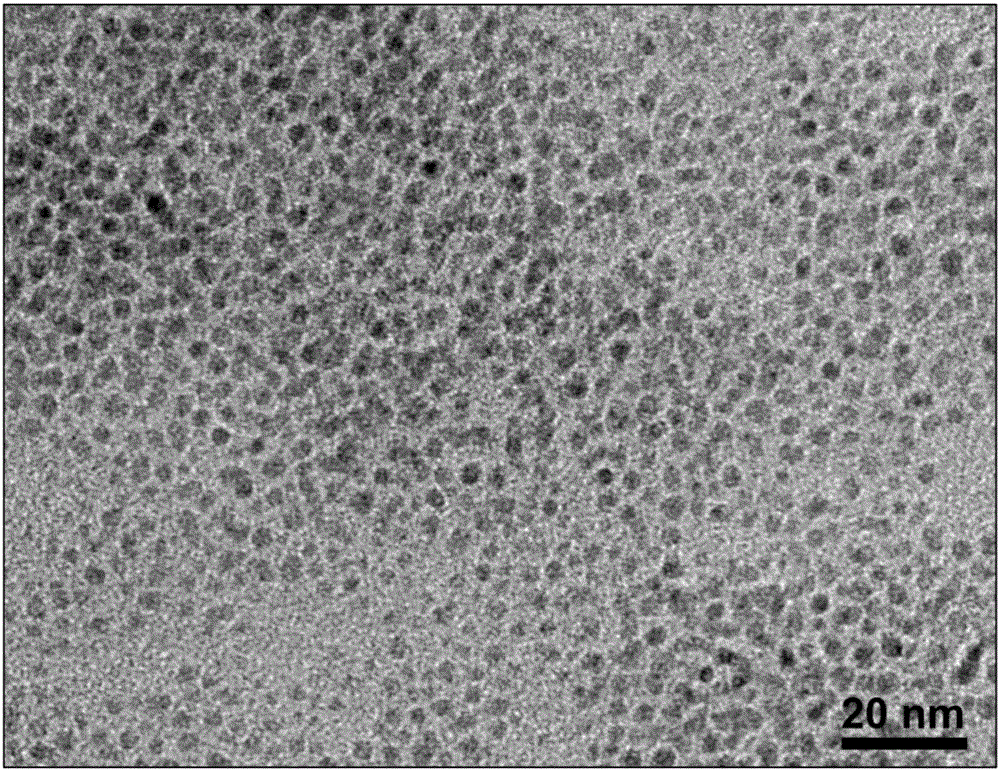
圖2為本發明實施例1制得的黑磷量子點的低倍透射電鏡圖(TEM);
圖3為本發明實施例1制得的黑磷量子點的高分辨透射電鏡(HRTEM)圖;
圖4為本發明實施例1制得的黑磷量子點的拉曼光譜圖;
圖5為本發明實施例4制得的黑磷量子點在N-甲基吡咯烷酮溶液中的紫外-可見-近紅外吸收光譜圖;
圖6為本發明實施例4制得的黑磷量子點的低倍透射電鏡圖。
具體實施方式
以下所述是本發明實施例的優選實施方式,應當指出,對于本技術領域的普通技術人員來說,在不脫離本發明實施例原理的前提下,還可以做出若干改進和潤飾,這些改進和潤飾也視為本發明實施例的保護范圍。
下面分多個實施例對本發明實施例進行進一步的說明。本發明實施例不限定于以下的具體實施例。在不變主權利的范圍內,可以適當的進行變更實施。
本發明實施例中,所述黑磷塊體是購買自Smart-Elements公司。
實施例1
一種黑磷量子點的制備方法,包括如下步驟:
(1)在充氮氣的手套箱中稱取10mg的塊體黑磷,將其研磨并分散到10mL的N-甲基吡咯烷酮中,密封,得到1mg/mL的第一分散液;
(2)取上述第一分散液2mL,加入18mL的N-甲基吡咯烷酮進行稀釋,然后加入200mg的氫氧化鈉,攪拌均勻得到第二分散液;
(3)將上述第二分散液加入到聚四氟乙烯高壓反應釜中,在溫度為140℃中加熱反應18h,待所得反應液冷卻至室溫后,在7000轉/分鐘的轉速下離心20分鐘,收集上清液,得到分散在N-甲基吡咯烷酮中的黑磷量子點。
本實施例1所得黑磷量子點較穩定,可以在室溫下存放1個月。將本實施例1所得黑磷量子點進行表征,結果如圖1~圖4所示,其中,圖1是黑磷量子點在N-甲基吡咯烷酮溶液中的紫外-可見-近紅外吸收光譜圖;圖2是黑磷量子點的低倍透射電鏡圖;圖3是黑磷量子點的高分辨透射電鏡圖;圖4是黑磷量子點的拉曼光譜圖。
從圖1中可以看出:黑磷量子點在300-900nm區域表現出明顯的吸收特性,其吸收峰值約在500nm,這與黑磷量子點的尺寸較小時,其帶隙有所增加有關系;圖2中可以看出所制得的黑磷量子點的單分散性好、尺寸均一,其尺寸在3nm左右;從圖3中可以明顯的看到黑磷量子點原子層的晶格條紋,其中,晶格間距為0.21nm對應于黑磷晶體的(014)晶面;從圖4中可以看出黑磷量子點的拉曼光譜圖相對于塊體黑磷有明顯的藍移(向右移動),證明了黑磷量子點比較薄。
實施例2
一種黑磷量子點的制備方法,包括如下步驟:
在充氮氣的手套箱中稱取30mg的塊體黑磷,將其研磨并分散到25mL的N-甲基吡咯烷酮中,密封,得到1.2mg/mL的第一分散液;
取上述第一分散液4mL,加入36mL的N-甲基吡咯烷酮,另外加入400mg的氫氧化鈉,攪拌均勻得到第二分散液;
將上述第二分散液加入到高壓反應釜中,在溫度為120℃中加熱反應14h,加熱完畢、待體系冷卻至室溫后,對所得反應液在9000轉/分鐘的轉速下離心10分鐘,收集上清液;再對收集的上清液進行高速離心20min,轉速為12000rpm,收集固體產物或將所述固體產物分散到無水乙醇中,得到分散在無水乙醇中、尺寸為4nm的黑磷量子點。
實施例3
一種黑磷量子點的制備方法,包括如下步驟:
在充氮氣的手套箱中稱取12mg的塊體黑磷,將其研磨并分散到20mL的N-甲基吡咯烷酮中,密封,得到0.6mg/mL的第一分散液;
取上述第一分散液3mL,加入27mL的二甲基甲酰胺,另外加入300mg的氫氧化鈉,攪拌均勻得到第二分散液;
將上述第二分散液加入到高壓反應釜中,在溫度為130℃中加熱反應10h,加熱完畢、待體系冷卻至室溫后,對所得反應液在6000轉/分鐘的轉速下離心30分鐘,收集上清液,得到分散在N-甲基吡咯烷酮中的黑磷量子點。
實施例4
一種黑磷量子點的制備方法,包括如下步驟:
在充氮氣的手套箱中稱取25mg的塊體黑磷,將其研磨并分散到25mL的N-甲基吡咯烷酮中,密封,得到1mg/mL的第一分散液;
取上述第一分散液2mL,加入18mL的N-甲基吡咯烷酮,另外加入200mg的氫氧化鈉,攪拌均勻得到第二分散液;
將上述第二分散液加入到三口燒瓶中,在溫度為140℃下加熱攪拌6h,其中攪拌的速度為4000轉/分;將體系冷卻至室溫后,對所得反應液在7000轉/分鐘的轉速下離心20分鐘,收集上清液,得到分散在N-甲基吡咯烷酮中的黑磷量子點。
將本實施例4所得的黑磷量子點進行表征,結果如圖5~圖6所示,其中,圖5是黑磷量子點在N-甲基吡咯烷酮溶液中的紫外-可見-近紅外吸收光譜圖;圖6為本發明實施例4制得的黑磷量子點的低倍透射電鏡圖。
從圖5中可以看出:黑磷量子點在300-900nm區域表現出明顯的吸收特性,從圖6中可以看出黑磷量子點的尺寸均一,分散性好,其尺寸在2.6nm左右。
此外,與同等條件下,采用高壓反應釜制得的黑磷量子點(3nm)相比,本實施例4制得的黑磷量子點的粒徑略小,這可能是還采用外力攪拌所致。
實施例5
在充氮氣的手套箱中稱取30mg的塊體黑磷,將其研磨并分散到15mL的N-甲基吡咯烷酮中,密封,得到1.2mg/mL的第一分散液;
取上述第一分散液3mL,加入27mL的N-甲基吡咯烷酮,另外加入300mg的氫氧化鈉,攪拌均勻得到第二分散液;
將上述第二分散液加入到三口燒瓶中,在溫度為120℃下加熱攪拌10h,其中攪拌的速度為3500轉/分;待反應體系冷卻至室溫后,對所得反應液在6000轉/分鐘的轉速下離心25分鐘,收集上清液,得到分散在N-甲基吡咯烷酮中的黑磷量子點。
實施例6
在充氮氣的手套箱中稱取12mg的塊體黑磷,將其研磨并分散到20mL的N-甲基吡咯烷酮中,密封,得到0.6mg/mL的第一分散液;
取上述第一分散液4mL,加入36mL的N-甲基吡咯烷酮,另外加入400mg的氫氧化鈉,攪拌均勻得到第二分散液;
將上述第二分散液加入到三口燒瓶中,在溫度為160℃下加熱攪拌8h,其中攪拌的速度為3000轉/分;然后將反應體系冷卻至室溫后,對所得反應液在9000轉/分鐘轉速下離心15分鐘,取上清液,得到分散在N-甲基吡咯烷酮中、尺寸為3.5nm的黑磷量子點。
以上所述僅為本發明的較佳實施例而已,并不用以限制本發明,凡在本發明的精神和原則之內所作的任何修改、等同替換和改進等,均應包含在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!