可重復利用工藝水的水性粘合劑組合物及利用其來粘合纖維狀材料的方法與流程
本發明涉及一種可重復利用工藝水的水性粘合劑組合物及使用其來粘合纖維狀材料的方法,更詳細地,涉及一種包含一種以上的還原糖、一種以上的氨基酸,以及一種以上的微生物最小抑制濃度(Minimum In hibitory Concentration,MIC)為1%以下的醛類化合物的水性粘合劑組合物及使用其來粘合纖維狀材料的方法。本發明的粘合劑組合物與現有的酚醛樹脂(PFR)相比,價格低廉且能夠顯示出同等以上的物理性質,最終產品不含有或者基本上不會釋放出甲醛或苯酚等有毒物質,在加工時能夠減少作為現有PFR樹脂的缺陷的臭氣問題,與不含有甲醛或苯酚等有毒物質的現有的環保樹脂相比,能夠顯著改善加工后的產品的耐水性和拉伸強度、硬度等機械特性及粉塵率,而且因粘合劑溶液具有防腐性能,從而能夠重復利用在使用粘合劑成分的纖維狀材料的粘合工序中所產生的工藝水,而不會引起因微生物導致的腐敗,因此具有經濟性。
背景技術:
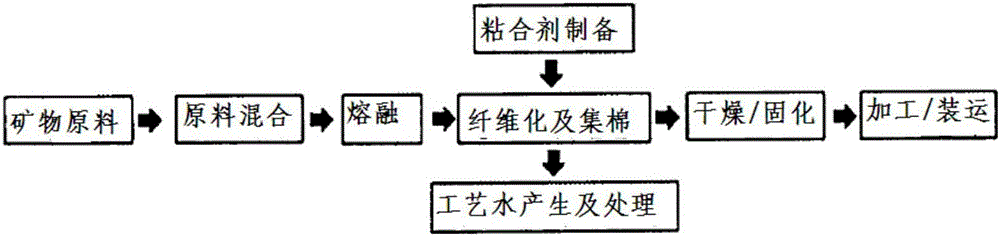
在圖1中概略地示出了使礦物纖維化,并對其進行加工而產品化的玻璃棉及巖棉等的制備工序。
即,礦物原料混合物在高溫下熔融且纖維化,并以涂布有粘合劑的狀態集棉。尤其是在涂布粘合劑并集棉的工序中,約有15~35%左右的粘合劑溶液會流到設備的側面或地面上,利用沖洗水來清洗該粘合劑溶液。此外,其它的制備設備也會被礦物纖維污染,利用沖洗水并采用相同的方法進行去除。本說明書中將這種在制備工序中產生的、含有粘合劑成分的沖洗水稱為“工藝水。”對于所述工藝水,利用過濾器來去除包含在所述工藝水中的纖維狀材料,并收集到集水槽中,然后再將其與粘合劑溶液混合或使用物理、化學、生物學方法等,將其中所含的有機物分解至不會腐敗的濃度以下。對于前者而言,具有可以重復利用損失的粘合劑成分的優點,但存在需要控制不讓其腐敗的問題。
目前,作為用于纖維狀材料的粘合的粘合劑正在使用的主要是價格低廉且物理性質優異的PFR。將PFR用作粘合劑時,所產生的工藝水含有苯酚或甲醛等毒性物質,因此具有可以重復利用而不會引起腐敗問題的優點。但是,PFR在制備工序中,甚至在施工后也會釋放出作為致癌物質的甲醛,而且苯酚也是毒性物質,其未反應物會持續地泄露到外部環境中,由此會帶來不良影響。
韓國授權專利第240044號、美國公開專利第2012-0133073號、韓國公開專利第1999-0037115號、韓國公開專利第2008-0049012號及韓國公開專利第2010-0065273號等中公開了以將源自天然物質的環保物質作為粘合劑成分使用作為特征的組合物。但是,上述文獻中記載的技術中,固化物的物理性質與現有的PFR樹脂相比要差,而且粘合劑溶液本身幾乎沒有防腐效果,因此只能將所產生的工藝水中的有機粘合劑成分進行分解或利用其它特殊的設備和工序來進行重復利用。
技術實現要素:
要解決的技術問題
本發明是為了解決上述現有技術中存在的問題而提出的,本發明要解決的技術問題為,提供一種水性粘合劑組合物及使用其來粘合纖維狀材料的方法,本發明的水性粘合劑組合物在玻璃棉及巖棉等纖維狀材料的粘合中,與現有的酚醛樹脂相比,價格低廉且能夠顯示出同等以上的物理性質,并且不含有或者基本上不會釋放出甲醛或苯酚等有毒物質,在加工時能夠減少作為現有PFR樹脂的缺陷的臭氣問題,并且能夠重復利用在纖維狀材料的粘合工序中所產生的工藝水,而不會引起因微生物導致的腐敗,因此具有經濟性,而且能夠顯著改善加工后的產品的耐水性和拉伸強度、硬度等機械特性及粉塵率。
技術方案
為了實現上述目的,本發明提供一種包含一種以上的還原糖、一種以上的氨基酸,以及一種以上的微生物最小抑制濃度(Minimum Inhibit ory Concentration,MIC)為1%以下的醛類化合物的水性粘合劑組合物。
根據本發明的另一方面,提供一種纖維狀材料的粘合方法,所述粘合方法包括以下步驟:將所述水性熱固性粘合劑組合物噴射到纖維狀材料上;以及對噴射的粘合劑組合物進行熱固化。
發明的效果
使用本發明的粘合劑組合物來粘合的纖維狀材料產品與現有的酚醛樹脂相比,價格低廉且能夠顯示出同等以上的物理性質,不含有或者基本上不會釋放出甲醛或苯酚等有毒物質,在加工時能夠減少作為現有PFR樹脂的缺陷的臭氣問題。此外,與現有的環保粘合劑不同,粘合劑溶液本身具有防腐性能,因此能夠重復利用在使用粘合劑成分粘合纖維狀材料的工序中所產生的工藝水,而不會引起因微生物導致的腐敗,因此具有經濟性,而且能夠顯著改善加工后的產品的耐水性和拉伸強度、硬度等機械特性及粉塵率。
附圖說明
圖1概略地示出了使礦物纖維化并對其進行加工而產品化的制備工序。
優選實施方式
下面,對本發明進行更加詳細說明。
1912年法國化學家美拉德(Louis-Camille Maillard)用存在于食品中的糖類與氨基酸之間的反應(以下稱為“美拉德反應”)來說明了在高溫下食品中所產生的非酶棕色化反應(“Action of Amino Acids on Suga rds.Formation of Melanoidins in a Methodical Way”,Compt.Rend.,1 54:66)。此外,1953年美國化學家霍奇(John E.Hodge)在論文中詳細記載了上述反應的實施機理(“Chemistry of Browing Reactions in Mo del Systems”,J.Agric.Food Chem.,1953,1,928-943)。即,說明了以下內容:在高溫下,帶有反應性醛基的醛糖形態的糖與氨基酸進行反應,從而形成N-取代葡基胺的中間體,通過阿馬多里重排以多種途徑進行反應,從而使得多種低分子量揮發性有機物分解出來,同時形成被稱為蛋白黑素的褐色的含氮高分子和其共聚物。尤其是在1966年魯尼(Ll oyd W.Rooney)和塞倫(Ali Salem)在他們的博士學位論文中報道了以下內容:將還原糖和氨基酸水溶液進行高溫反應(在水溶液狀態下于95℃下進行12小時,與淀粉混合后于268℃下進行30分鐘)時,通過觀察顏色變化及測定分解出的低分子量有機化合物(甲醛、乙醛、丙酮、異丁醛、異戊醛)的量的方法來確定美拉德反應的進行程度的情況下,葡萄糖或木糖等還原糖與蛋氨酸、色氨酸、苯丙氨酸、賴氨酸、組氨酸、甘氨酸、丙氨酸、纈氨酸、亮氨酸、異亮氨酸等氨基酸活躍地進行反應(“Studies of the Carbonyl Compounds Produced by Sugar-Amino Acid Reactions.I.Model Systems,Ph.D Thesis,the Graduate Faculty of Kansas State University,1966,No.559,539-550)。
已知為了增強食品的香氣和味道,在食品領域中大量利用美拉德反應。但是,美拉德反應在粘合劑領域中一直沒有被準確地解釋。例如,由糖類和氨基酸的高溫反應所形成的、被稱為蛋白黑素的褐色的含氮高分子可以通過在高溫下對糖蜜進行加熱來容易地獲得,其中所述糖蜜作為有機粘合劑而經常使用。糖蜜為純化糖類時所產生的副產物,其為以質量比包含約50%的含有還原糖的糖類,包含約5~10%的含有氮元素的化合物(蛋白質、氨基酸類、低聚物類)等的混合物,在高溫下,其組成成分中的還原糖與蛋白質/氨基酸進行反應,從而形成蛋白黑素。
美國專利第3,961,081號中公開了在家畜的飼料中,將糖蜜作為粘合劑使用的內容。在此,記載了將含有糖蜜的組合物于105~155℃的高溫下施加真空而去除縮合物,從而可以制備成包含蛋白黑素的、堅硬形態的粉末。
美國專利第5,416,139號中公開了在制備建筑結構材料時,將糖蜜作為粘合劑使用的內容。在此,記載了將含有糖蜜的組合物于150~180℃的高溫下進行射出,從而能夠制得包含蛋白黑素的、經過固化的復合材料。
韓國申請專利第1987-0005710號中記載了使用酚醛樹脂(PFR)或氨基甲醛樹脂的一部分與糖類(糖蜜、葡聚糖、葡萄糖、果糖、蔗糖等)及木質素磺酸鹽的混合物可以降低氨基樹脂及苯酚樹脂的甲醛釋放量。在此,具有還原糖的成分和含氨基的成分的組合物在高溫下能夠形成蛋白黑素。
韓國公開專利第10-2008-0049012號中記載了聚羧酸的銨鹽與還原糖進行美拉德反應,從而能夠用作粘合劑。
美國專利第4,524,164號中記載了用于鋸屑等木質纖維素材料的熱固性粘合劑的組成及應用方法,在此,采用以下方法:在金屬催化劑存在的條件下,于水溶液狀態下,使包含還原糖的糖類(乳糖、麥芽糖、葡萄糖、半乳糖、蔗糖、直鏈淀粉、支鏈淀粉、葡聚糖、糖蜜、乳漿)與尿素等固化劑及銨鹽在50~200℃下反應30分鐘~18小時,并將其與有機酸酐和鋸屑等木質纖維素原料進行混合后在高溫下進行加工。在此,還原糖和尿素、銨鹽及有機酸酐在高溫下能夠形成蛋白黑素。
法國專利第2,924,719號中記載了使用由單糖類、多糖類(葡聚糖或糖蜜)、聚羧酸及催化劑組成的水性粘合劑組合物能夠將巖棉或玻璃棉制成保溫材料產品的內容,所述粘合劑組合物也能夠在高溫下形成蛋白黑素,從而對纖維進行粘合。
本發明如前面所述,將本領域公知的還原糖與氨基酸在高溫下進行美拉德反應而獲得的蛋白黑素用作粘合劑。此外,為了對制備纖維狀集合體的工序中所產生的、包含粘合劑成分的工藝水賦予防腐力而進行重復利用,以及為了提高蛋白黑素粘合劑的性能,本發明的特征為,將微生物最小抑制濃度(Minimum Inhibitory Concentration,MIC)為1%以下的醛類化合物用于粘合劑組合物。
本發明中使用的微生物最小抑制濃度(MIC)為1%以下的醛類化合物可以通過下述方法來進行選擇。
將本發明中使用的還原糖和氨基酸的重量比調節為1/1,從而制成5%水溶液,并在常溫下放置1周使其腐敗。將從腐敗的試料中分離的污染菌株用作試驗菌株,并分別接種到作為篩選對象的各醛稀釋液中,并于35℃下在培養箱中培養的同時,觀察微生物的增殖及死亡現象,從而篩選出MIC為1%以下的醛類化合物。
本發明中使用的MIC為1%以下的醛類化合物作用于微生物的細胞壁或細胞質而打亂它們的作用,或者與硫醇基、氨基、巰基等特性基團發生化學反應來破壞微生物,由此能夠抑制微生物的增殖或殺滅微生物。
此外,所述醛類化合物除了具有對水性粘合劑組合物賦予防腐力的作用以外,還能夠與本發明中使用的氨基酸的氨基結合,從而在高溫固化時能夠形成物理性質優異的三維交聯的高分子網狀結構。
因此,本發明的水性粘合劑組合物在涂布到纖維化的礦物上,并在烘箱中高溫固化之前,雖然有可能會包含有毒性醛類化合物(例如,甲醛),但是在高溫固化后獲得的最終絕熱材料產品中實際上不會包含未反應的或在固化反應時分解而產生的有毒性醛類化合物。
即,就使用本發明的水性粘合劑組合物制備的絕熱材料產品而言,通過小型腔室法(KSM ISO16000及KSM1998)測定的甲醛的釋放量限為0~0.005mg/m3·hr范圍,顯示出0至幾乎為0的數值。
此外,本發明中使用的醛類化合物與其它防腐劑不同,還具有不會誘發金屬的腐蝕或在產品中殘留有害的鹵素元素或苯酚等化合物的優點。
本發明的粘合劑組合物包含碳原子數為3以上的還原糖。本發明中,還原糖是指含有醛或通過異構化能夠具有醛結構的醛糖或酮糖糖類,具體地,可以單獨或組合使用葡萄糖、麥芽糖、果糖、半乳糖、乳糖、纖維二糖、龍膽二糖、蕓香糖、甘油醛等單糖類及二糖類,但并不限定于此。
以還原糖和氨基酸的合為100重量份計,本發明的粘合劑組合物中所包含的還原糖的量優選為40~95重量份,更優選為60~90重量份,進一步優選為70~90重量份。以還原糖和氨基酸的合為100重量份計,當粘合劑組合物內的還原糖的含量不足40重量份時,由粘合劑組合物形成的固化物的硬度會降低,當該含量超過95重量份時,組合物的穩定性及固化物的固化度會降低。
本發明的粘合劑組合物包含一種以上的氨基酸。本發明中的氨基酸為在一個分子中分別包含一個以上的氨基和羧基的化合物,具體地,可以單獨或組合使用甘氨酸、丙氨酸、纈氨酸、亮氨酸、異亮氨酸、蘇氨酸、絲氨酸、半胱氨酸、蛋氨酸、天冬氨酸、天冬酰胺、谷氨酸、二碘酪氨酸、賴氨酸、精氨酸、組氨酸、苯丙氨酸、酪氨酸、色氨酸、脯氨酸、羥脯氨酸、谷氨酰胺等,但并不限定于此。
以還原糖和氨基酸的合為100重量份計,本發明的粘合劑組合物中包含的氨基酸的量優選為5~60重量份,更優選為10~40重量份,進一步優選為10~30重量份。以還原糖和氨基酸的合為100重量份計,當粘合劑組合物內的氨基酸含量不足5重量份時,組合物的穩定性及固化物的固化度會降低,當該含量超過60重量份時,由粘合劑組合物形成的固化物的硬度會降低。
根據本發明的一具體例,為了提高氨基酸對于水的溶解度,優選使用氨基的一部分或全部(例如,包含于氨基酸中的氨基的20~100當量%,優選為30~100當量%)被酸中和的氨基酸。用于中和氨基酸所使用的酸,可以例舉如硫酸、磷酸、羧酸、有機磺酸等,其形態可以不受限制地使用單分子、二聚物、三聚物、低聚物或高分子形態的酸化合物。根據情況也可以使用被胺化合物或氨中和的形態。優選地,可以使用在后述的常溫及常壓(即,25℃,1個大氣壓)條件下的沸點為300℃以上(例如,300~500℃)的高沸點酸化合物。
本發明的粘合劑組合物包含一種以上的MIC為1%以下的醛類化合物。本發明中的MIC為1%以下的醛類化合物可以單獨或組合使用戊二醛、甲醛、乙二醛等,但并不限定于此。
以還原糖和氨基酸的合為100重量份計,本發明的粘合劑組合物中包含的MIC為1%以下的醛類化合物的量優選為0.01~10重量份,優選為0.1~5重量份。以還原糖和氨基酸的合為100重量份計,當粘合劑組合物內的所述醛類化合物含量不足0.01重量份時,由于防腐力不足,會使得產生的包含粘合劑成分的工藝水容易因腐敗而變質,并誘發惡臭,當該含量超過10重量份時,會引起固化物的過度固化,從而使得涂膜容易破碎。
作為本發明的主要成分的還原糖和氨基酸是取自植物的淀粉、糖蜜等經過水解或發酵工序而獲得的,無需擔心資源的枯竭,因此能夠使制備和廢棄時所產生的二氧化碳的量最小化。此外,產品中幾乎不含有苯酚、甲醛等有毒物質(其是指在甲醛的情況下,用小型腔室法測定釋放量時,顯示0~0.005mg/m3·hr范圍的0至幾乎為0的數值)。
優選地,本發明的粘合劑組合物可以進一步包含在常溫及常壓(即,25℃,1個大氣壓)條件下的沸點為300℃以上(例如,300~500℃)的高沸點酸化合物。這種高沸點酸化合物可以提高氨基酸對于水的溶解度,并且在噴射到纖維狀集合體上之后,在高溫烘烤工序中為了提高固化反應速度而使用,在高溫固化反應時為了防止揮發,優選地,該高沸點酸化合物在常溫及常壓條件下的沸點為300℃以上。作為高沸點酸化合物的例子,可以例舉如硫酸、磷酸、羧酸、有機磺酸等,其形態可以不受限制地使用單分子、二聚物、三聚物、低聚物或高分子形態的酸化合物。根據情況也可以使用被胺化合物或氨中和的形態
本發明的粘合劑組合物中包含高沸點酸化合物的情況下,以還原糖和氨基酸的合為100重量份計,所述高沸點酸化合物的量可以優選為0.1~10重量份,更優選為0.1~5重量份,進一步優選為0.1~3重量份。以還原糖和氨基酸的合為100重量份計,當粘合劑組合物內的高沸點酸化合物的含量不足0.1重量份時,會發生未固化,從而會降低固化物的物理性質,當該含量超過10重量份時,會產生易碎的固化物。
本發明的粘合劑組合物中,除了上述成分以外,在能夠實現本發明的目的的范圍內,可以根據需要選擇性地進一步包含一種以上的添加劑。作為有益的附加添加劑,可以例舉如用于提高纖維狀集合體的耐水性的防水劑、用于防止設備腐蝕的防銹劑、用于降低產品的粉塵發生率的防塵油、用于調節pH的緩沖劑、用于提高附著性的偶聯劑、用于提高固化物的機械物理性質的固化劑等,但并不限定于此,可以使用本領域中通常使用的添加劑。這種附加添加劑的使用量不受特別的限制,例如,以還原糖和氨基酸的合為100重量份計,可以以0.1~10重量份的范圍使用各個添加劑,但并不限定于此。
此外,在本發明中,為了防止以數%包含在制備工序中產生的有機粘合劑成分的工藝水的腐敗,可以進一步添加防腐劑。這種防腐劑可以在制備粘合劑時射入到混合槽中,或者可以加入到工藝水集水槽、傳輸管道、水處理設備等任何設備中,并且只要是本領域中通常使用的防腐劑,則可以不受種類的限制而使用。相對于所產生的工藝水的重量,防腐劑的添加量優選可以使用500~5,000ppm,更優選可以使用1,000~3,000ppm的范圍。為了殺滅微生物,根據情況可以在常壓及55~100℃的溫度范圍內對粘合劑混合槽進行一定時間以上的加熱。就微生物的死亡或微生物的濃度的確認而言,可以通過使用本領域中通常使用的診斷試劑盒和微生物培養設備來容易地進行確認。
就本發明的粘合劑組合物而言,作為用于將上述成分均勻地涂布到纖維狀材料上的稀釋劑,可以使用水(工業用水、地下水、工藝水等),并根據需要可以將其固含量調節為2~50重量%,優選為5~20重量%(即,以組合物總100重量%計,水的含量為50~98重量%,優選為80~95重量%)。當作為稀釋劑的水的量過多時,會消耗過多的用于揮發水的能量,另一方面,當該含量過少時,粘合劑組合物不能很好地涂布到纖維狀材料上,并且會導致最終產品內的粘合劑含量不必要地增加,因此不優選。
就本發明的粘合劑組合物而言,例如在120℃以上的溫度下進行熱處理的情況下,會引起還原糖的醛基和氨基酸的氨基的阿馬多里(Amador i)中間體中發生的美拉德(Maillard)反應、氨基酸的羧酸基和氨基的自身酰胺化反應、還原糖的羥基和氨基酸的羧酸基的酯化反應、金屬離子和還原糖或氨基酸化合物中存在的氮、氧、硫、鹵素原子的配位鍵結合等多種固化反應,由此會形成水不溶性的高分子,因此可以作為耐水性等物理性質非常優異的粘合劑來使用。
因此,本發明的另一方面提供一種纖維狀材料的粘合方法及使用本發明的水性熱固性粘合劑組合物來粘合的纖維狀材料,其中所述粘合方法包括以下步驟:將本發明的水性熱固性粘合劑組合物噴射到纖維狀材料上;以及對噴射的粘合劑組合物進行熱固化。。
就本發明的纖維狀材料的粘合方法而言,所述水性熱固性粘合劑組合物以未固化的水溶液或水分散液狀態被噴射到纖維狀材料上。作為所述纖維狀材料的例子,可以例舉如無機質纖維(例如巖棉、玻璃棉、陶瓷纖維等)或天然樹脂及合成樹脂中獲得的纖維等的單纖維集合體,但并不限定于此。
然后,對噴射有粘合劑組合物的纖維狀材料集合體進行熱處理,從而使粘合劑組合物熱固化。用于固化的熱處理溫度適當為120℃以上(例如,120~300℃,優選為150~250℃)。當熱處理溫度過低時,會發生未固化,另一方面,當該溫度過高時,會發生過度固化,由此產生的粉塵會成為問題。
以經過粘合的纖維狀材料總100重量份計,根據本發明制備的經過粘合的纖維狀材料內的固化的粘合劑含量例如可以為2~15重量%。
下面,將通過實施例對本發明進行更加具體地說明。但是,這些實施例只是為了有助于理解本發明,本發明的范圍從任何意義上來說都不能被這些實施例所限定。
[實施例]
實施例1
將370kg的葡萄糖(D-glucose)(固含量:91重量%)、70kg的谷氨酰胺(glutamine)(固含量:98重量%)、2.5kg的硫酸銨(ammonium sulfate)(固含量:98重量%)、1.5kg的甲醛(福爾馬林,UNID公司)(固含量:37重量%)、3kg的防塵油(Dust Oil,Govi-Garo217S)、2kg的硅類防水劑(KCC-SI1460Z)及4050kg的蒸餾水加入到混合容器中攪拌30分鐘后,利用氨水將pH調至8~9,進一步攪拌10分鐘,從而制得粘合劑組合物。制得的粘合劑組合物的固含量約為9重量%。
實施例2
將370kg的葡萄糖(D-glucose)(固含量:91重量%)、70kg的谷氨酰胺(glutamine)(固含量:98重量%)、2.5kg的硫酸銨(ammonium sulfate)(固含量:98重量%)、660kg的蒸餾水加入到在反應器外部設置有可以使用蒸汽及冷卻水來調節溫度的夾套(jacket)的反應器中,并升溫至85℃后使反應物的溫度維持3小時。在此,將3390kg的室溫的蒸餾水、1.5kg的甲醛(福爾馬林,UNID公司)(固含量:37重量%)、3kg的防塵油(Dust Oil,Govi-Garo217S)及2kg的硅類防水劑(KCC-SI1460Z)加入到混合容器中攪拌30分鐘后,利用氨水將pH調至8~9,進一步攪拌10分鐘,從而制得粘合劑組合物。制得的粘合劑組合物的固含量約為9重量%。
實施例3
除了使用1.5kg的乙二醛(Glyoxal solution)(固含量:40重量%)來代替甲醛之外,采用與實施例1相同的方法制備粘合劑組合物。制得的粘合劑組合物的固含量約為9重量%。
實施例4
除了用1.5kg的戊二醛(Glutaraldehyde)(固含量:50重量%)代替甲醛之外,采用與實施例1相同的方法制備粘合劑組合物。制得的粘合劑組合物的固含量約為9重量%。
比較例1:使用苯酚/甲醛樹脂來制備粘合劑
將404kg的苯酚/甲醛樹脂(金剛高麗化學(KCC)制備)、3800kg的蒸餾水、2kg的防水劑(SI1460Z-KCC)及3kg的防塵油(Garo217S)加入到混合容器中,用攪拌器攪拌30分鐘,從而制得粘合劑組合物。制得的粘合劑組合物的固含量約為9重量%。
比較例2:使用聚羧酸來制備粘合劑(韓國公開專利第2008-0049012號的實施例1)
將158kg的葡萄糖(D-glucose)(固含量:91重量%)、44kg的檸檬酸(citric acid)(固含量:98重量%)、50kg的氨水(25%)、3750kg的蒸餾水、2kg的防水劑(SI1460Z-KCC)及3kg的防塵油(Garo217S)加入到混合容器中,用攪拌器攪拌30分鐘,從而制得粘合劑組合物。制得的粘合劑組合物的固含量約為4.5重量%。
比較例3:制備不含有MIC為1%以下的醛類化合物的還原糖/氨基酸粘合劑
將370kg的葡萄糖(D-glucose)(固含量:91重量%)、67kg的谷氨酰胺(glutamine)(固含量:98重量%)、2.5kg的硫酸銨(ammonium sulfate)(固含量:98重量%)、3kg的防塵油(Dust Oil,Govi-Garo217S)、2kg的硅類防水劑(KCC-SI1460Z)及4050kg的蒸餾水加入到混合容器中,用攪拌器攪拌30分鐘,從而制得粘合劑組合物。制得的粘合劑組合物的固含量約為9重量%。
實驗例:使用粘合劑的纖維狀材料的粘合
使高溫的熔融玻璃通過紡紗機,以每小時2100kg/hr的速度進行纖維化,同時向落到集棉室中的玻璃纖維,以47L/分鐘的量分別噴射上述實施例1至實施例4及比較例1至比較例3中制得的粘合劑組合物后,經過干燥工序獲得玻璃棉絕熱材料。在比較例3的粘合劑的情況下,由于析出了氫氧化鈣,因此將粘合劑溶液用150目的不銹鋼過濾器過濾掉塊狀物后進行了噴射,其它的工序采用與其它的例子相同的方式實施。
對按照上述方法制得的各個玻璃棉實施以下各項實驗。
耐水性
準備100mm(長)×100mm(寬)×50mm(厚度)的玻璃棉樣品后,實施標準化的48小時的耐水性試驗。使樣品漂浮在裝有純水的燒杯中,測定完全下沉為止所用的時間。耐水性等級越接近0則表示耐水性越差,越接近5則表示越優異。在實施例1、2、3、4和比較例1、2的情況下,樣品在水上漂浮72小時以上,而另一方面,在比較例3的情況下,樣品在24小時內下沉。實驗結果如下述表1中所示。
浸水性
準備100mm(長)×100mm(寬)×50mm(厚度)的玻璃棉樣品后,實施標準化的72小時的浸水性試驗。使樣品完全浸入裝滿純水的2L的燒杯中,并測定72小時的顏色變化。此時,客觀地將根據濃度的顏色變化程度進行數值化。浸水性等級越接近0表示浸水性越差,越接近5則表示越優異。在實施例1~4的情況下,72小時以上幾乎沒有出現顏色變化,而另一方面,在比較例3的情況下,在幾個小時內出現了嚴重的顏色變化。實驗結果如下述表1中所示。
甲醛釋放量
按照小型腔室法將試片放置在小型腔室后,捕集第7天的腔室中的空氣,利用高效液相色譜法(HPLC)分析捕集的空氣。具體的試驗方法采用了空氣凈化協會設定的方法,在第7天進行了結果判定。實驗結果如下述表1中所示。
拉伸強度
以4cm寬度大小分別制作了3個測試試樣。將牽引桿放置在比試樣的長度短的位置,并將試樣呈水平地固定在牽引桿上后,使試樣以與支撐桿垂直的方式緊固在支撐桿上。將拉伸強度測試儀的速度設為15mm/分鐘,將荷重顯示置于0后啟動。待測試儀自動停止后,測定顯示的最大荷重,并算出平均值。實驗結果如下述表1中所示。
粉塵率
以1.5cm的寬度和10cm的長度大小分別制作了4個測試試樣。在測試前進行稱重,然后將試樣置于測試儀上,以1m/分鐘的速度向前后左右搖晃。總測試時間是以每個試樣測試10分鐘為基準,待測試儀自動停止后稱取試樣的重量。使用粉塵率=[(測試后稱取的重量/測試前稱取的重量)-1]*100的計算式算出粉塵率,并用%表示。使用實施例1~4的粘合劑制得的玻璃棉試樣的粉塵與使用比較例的粘合劑的情況相比,產生的粉塵少。實驗結果如下述表1中所示。
復原率
準備10m(長)×1m(寬)×0.05m(厚度)的玻璃棉樣品后,將其卷成卷(Roll)狀,在常溫下存放8周后將其打開恢復成原來的狀態,并確認厚度變化。實驗結果如下述表1中所示。
抗霉菌性
對于抗霉菌性,根據美國材料與試驗協會(ASTM)G21-09試驗方法觀察試片上的霉菌生長率1個月而進行測定,其結果如下述表1中所示。
工藝水防腐力
就使用各實施例及比較例的粘合劑的情況所對應的工藝水樣品而言,用設備沖洗水來稀釋各個水性粘合劑,從而制備固含量被調整為2~3重量%的樣品。為了確認各個例子的工藝水樣品的防腐力,對于混合細菌實施激發試驗。
所使用的菌株為作為革蘭氏陽性菌的枯草芽孢桿菌(Bacillus subtili s ATCC 6633)、金黃色葡萄球菌(Staphylococcus aureus ATCC 6538),以及作為革蘭氏陰性菌的大腸桿菌(Escherichia coli ATCC 9637)、綠膿桿菌(Pseudomonas aeruginosa ATCC 9027)。在預培養中,在營養瓊脂(Nutrient Agar)上接種上述4種細菌,并于33~37℃下培養24小時,然后與生理食鹽水混合,使得細菌濃度成為約1×106-7cfu/g(cfu:菌落形成單位),從而制得混合菌液。
在各個實施例及比較例的工藝水樣品100g中分別接種1ml的上述混合菌液。之后,分別取1g的經過接種的試樣,將其稀釋到9ml的磷酸鹽緩沖生理食鹽水(PBS緩沖液)溶液中,制得系列稀釋。在瓊脂培養基中分別注入100μl的上述稀釋液,并用滅菌棒均勻展開。然后將培養基倒置,并在33~37℃下培養48小時后計數。
對于判定,將菌死亡的情況設為0,將1×106-7cfu/g的嚴重濃度的情況設為4,并分成0~4的5個階段區間而決定(0:沒有細菌存活,1:污染的痕跡,2:輕度污染,3:中度污染,4:嚴重污染)。優選地,為了使工藝水具有防腐力,細菌在接種1周之內其菌數需要減少99.9%以上,并且在試驗期間不能發生增殖。其結果如表1中的工藝水防腐力項中所示。
表1
- 還沒有人留言評論。精彩留言會獲得點贊!