PslG在石油開采中的應用的制作方法
本發明涉及生物
技術領域:
中PslG在石油開采中的應用。
背景技術:
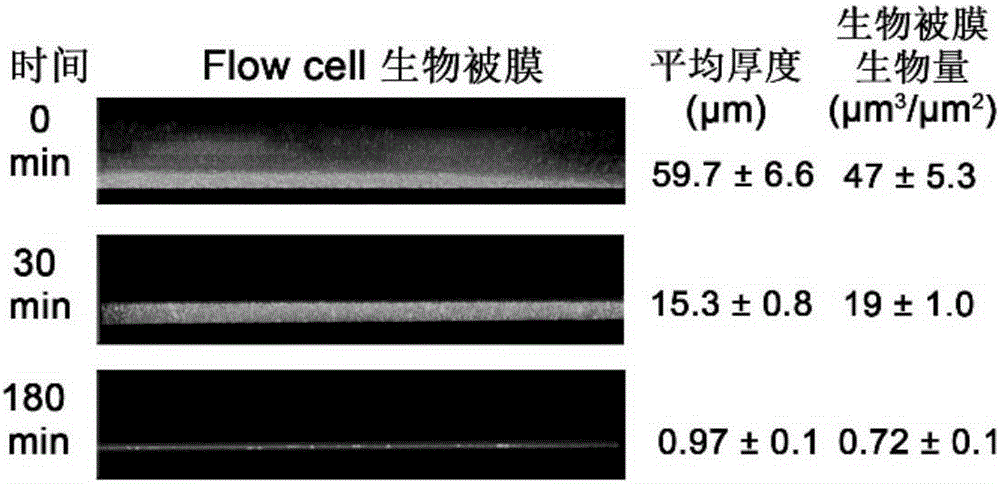
:微生物提高原油采收率技術(MicrobialEnhancedOilRecovery,MEOR)是指利用各種微生物(主要是細菌)及其代謝產物與油藏巖石、流體等發生生物化學作用,以提高原油采收率的技術。MEOR是繼聚合物驅、熱力驅、化學驅等方法之后新興的提高原油采收率的方法,亦稱微生物驅,該技術具有成本低、對油藏適應性強、不傷害地層和不污染環境等優點,被認為是二十一世紀最有發展前景的提高采油收率技術。至今,在世界上已有三十多個國家開展了相關的研究,并取得了顯著的效果。微生物提高原油采收率的機理十分復雜,其中微生物來源的表面活性劑以及基于微生物生物量的微生物調剖是最主要的作用機理。微生物生物量可以選擇性或非選擇性的封堵一些油層,改變巖石表面潤濕性,降低原油粘度,因此,對于原油采收率的提高非常重要。生物被膜(biofilm),也稱生物膜,是一種或多種微生物為適應自然環境而形成的膜性聚合體。根據《AnnuRevMicrobiol》等權威期刊所歸納發表的定義,生物被膜是指細菌粘附于接觸表面,分泌多糖基質、纖維蛋白、脂質蛋白等,將其自身包繞其中而形成的大量細菌聚集膜樣物。生物被膜由胞外多聚物包裹微生物細胞形成,能夠增強其中微生物對環境的抗逆性,使其更好的在各種環境中定殖。因此,油藏環境中生物被膜的形成有助于細菌生物量的積累,從而增強其生物調剖效果。然而,在某些環境條件下,過度形成的生物被膜以及細菌分泌的胞外多糖使微生物不能游離分散,導致部分原油的封堵,從而在一定程度上會降低采油效率。因此如果在驅油時利用有效降解胞外多糖及生物被膜的酶或試劑,可大大提高原油采收率。假單胞菌屬細菌可利用多種碳源生長,能產表面活性劑,因而廣泛存在于油藏環境。假單胞菌屬細菌也是微生物采油中用得最多的細菌。因此,可以有效瓦解或分散假單胞菌屬細菌形成的生物被膜或有效降解細菌分泌的胞外多糖的酶或試劑有應用于驅油過程并提高原油采收率的潛能。技術實現要素:本發明所要解決的技術問題是如何有效降解油藏環境中的胞外多糖并快速瓦解或分散油藏環境中的假單胞菌形成的生物被膜以提高原油采收率。為解決上述技術問題,本發明首先提供了PslG在石油開采中的應用。本發明所提供的PslG在石油開采中的應用中,PslG為如下A1)或A2)或A3):A1)氨基酸序列為序列2的蛋白質;A2)在序列2的氨基酸序列中經過取代和/或缺失和/或添加一個或幾個氨基酸殘基得到的具有相同功能的由A1)衍生的蛋白質;A3)在A1)或A2)的N端或/和C端連接標簽得到的融合蛋白質。為了使A1)中的蛋白質便于純化,可在序列表中序列1所示的蛋白質的氨基末端或羧基末端連接上如下表所示的標簽。表、標簽的序列標簽殘基序列Poly-Arg5-6(通常為5個)RRRRRPoly-His2-10(通常為6個)HHHHHHFLAG8DYKDDDDKStrep-tagII8WSHPQFEKc-myc10EQKLISEEDL上述A2)中的PslG可人工合成,也可先合成其編碼基因,再進行生物表達得到。上述A2)中的PslG的編碼基因可通過將序列表中序列1的所示的DNA序列中缺失一個或幾個氨基酸殘基的密碼子,和/或進行一個或幾個堿基對的錯義突變,和/或在其5′端和/或3′端連上表1所示的標簽的編碼序列得到。為解決上述技術問題,本發明還提供了與PslG相關的生物材料在石油開采中的應用。本發明所提供的與PslG相關的生物材料在石油開采中的應用中,所述生物材料為下述B1)至B8)中的任一種:B1)編碼PslG的核酸分子;B2)含有B1)所述核酸分子的表達盒;B3)含有B1)所述核酸分子的重組載體;B4)含有B2)所述表達盒的重組載體;B5)含有B1)所述核酸分子的重組微生物;B6)含有B2)所述表達盒的重組微生物;B7)含有B3)所述重組載體的重組微生物;B8)含有B4)所述重組載體的重組微生物。上述應用中,B1)所述核酸分子為如下b1)或b2)或b3):b1)核苷酸序列是序列表中序列2的cDNA分子或DNA分子;b2)與b1)限定的核苷酸序列具有75%或75%以上同一性,且編碼PslG的cDNA分子或DNA分子;b3)在嚴格條件下與b1)限定的核苷酸序列雜交,且編碼PslG的cDNA分子或DNA分子。其中,所述核酸分子可以是DNA,如cDNA、基因組DNA或重組DNA;所述核酸分子也可以是RNA,如mRNA或hnRNA等。序列1的所示的DNA分子編碼序列2所示的PslG。本領域普通技術人員可以很容易地采用已知的方法,例如定向進化和點突變的方法,對本發明的編碼PslG的核苷酸序列進行突變。那些經過人工修飾的,具有與本發明分離得到的PslG的核苷酸序列75%或者更高同一性的核苷酸,只要編碼PslG且具有PslG功能,均是衍生于本發明的核苷酸序列并且等同于本發明的序列。這里使用的術語“同一性”指與天然核酸序列的序列相似性。“同一性”包括與本發明的編碼PslG所示的氨基酸序列組成的蛋白質的核苷酸序列具有75%或更高,或85%或更高,或90%或更高,或95%或更高同一性的核苷酸序列。同一性可以用肉眼或計算機軟件進行評價。使用計算機軟件,兩個或多個序列之間的同一性可以用百分比(%)表示,其可以用來評價相關序列之間的同一性。上述應用中,所述嚴格條件是在2×SSC,0.1%SDS的溶液中,在68℃下雜交并洗膜2次,每次5min,又于0.5×SSC,0.1%SDS的溶液中,在68℃下雜交并洗膜2次,每次15min;或,0.1×SSPE(或0.1×SSC)、0.1%SDS的溶液中,65℃條件下雜交并洗膜。上述75%或75%以上同一性,可為80%、85%、90%或95%以上的同一性。上述應用中,B2)所述的含有編碼PslG的核酸分子的表達盒(PslG基因表達盒),是指能夠在宿主細胞中表達PslG的DNA,該DNA不但可包括啟動PslG基因轉錄的啟動子,還可包括終止PslG基因轉錄的終止子。進一步,所述表達盒還可包括增強子序列。可用現有的表達載體構建含有所述PslG基因表達盒的重組載體。上述應用中,所述載體可為質粒、黏粒、噬菌體或病毒載體。上述應用中,所述微生物可為酵母、細菌、藻或真菌。PslG的編碼基因可通過含有PslG的編碼基因的表達盒的重組載體導入微生物中得到重組微生物。為解決上述技術問題,本發明還提供了下述任一應用:C1)PslG或所述生物材料在制備石油開采產品中的應用;C2)PslG或所述生物材料在降解假單胞菌屬細菌生物被膜中的應用;C3)PslG或所述生物材料在制備降解假單胞菌屬細菌生物被膜產品中的應用;C4)PslG或所述生物材料在降解假單胞菌所產多糖中的應用;C5)PslG或所述生物材料在制備降解假單胞菌屬多糖產品中的應用;C6)PslG或所述生物材料在微生物提高原油采收率技術中的應用。為解決上述技術問題,本發明還提供了下述任一產品:D1)石油開采產品,其活性成分為PslG或所述生物材料;D2)降解假單胞菌屬細菌生物被膜產品,其活性成分為PslG或所述生物材料;D3)降解假單胞菌屬細菌生物被膜中多糖產品,其活性成分為PslG或所述生物材料。為解決上述技術問題,本發明還提供了假單胞菌屬細菌生物被膜的降解方法。本發明所提供的假單胞菌屬細菌生物被膜的降解方法,包括:在同一個反應體系中,將包被有生物被膜的假單胞菌屬細菌與PslG進行反應,降解所述假單胞菌屬細菌生物被膜。上述方法中,所述反應的溫度可為20-50℃,如25℃、30℃、37℃、40℃、45℃和50℃;和/或,所述反應的時間可為30min-180min,如30-150min。為解決上述技術問題,本發明還提供了下述任一應用:E1)所述產品在石油開采、降解假單胞菌屬細菌生物被膜或降解假單胞菌屬細菌生物被膜中多糖中的應用;E2)所述方法在石油開采中的應用;E3)所述產品在微生物提高原油采收率技術中的應用;E4)所述方法在微生物提高原油采收率技術中的應用。本發明中,所述多糖為可Psl多糖、Pel多糖、褐藻多糖和/或其他參與假單胞菌生物被膜形成的多糖。所述假單胞菌屬細菌可為銅綠假單胞菌、斯氏假單胞菌、惡臭假單胞菌、熒光假單胞菌、丁香假單胞菌等。本發明中,所述銅綠假單胞菌可為銅綠假單胞菌MSH3、MSH10、CIM51、CIM52、CIM53、PAO1、PAO1△psl菌株、PAO1PBAD-psl菌株、粘性菌株PDO300、MJK8菌株、MJK8△psl菌株或MJK8△pel菌株。所述斯氏假單胞菌可為斯氏假單胞菌A1501。實驗證明,本發明的來源于銅綠假單胞菌的PslG多糖水解酶,該酶可以在30分鐘內迅速將生物被膜瓦解分散為游離細菌,并在細菌可存活的條件下均具有活性。該酶瓦解分散生物被膜的原理在于其可以降解銅綠假單胞菌合成的名為Psl的胞外多糖。該多糖被稱為分子膠,可以促進細菌細胞的粘附和聚集。Psl胞外多糖是PslG的最佳底物,但該酶對假單胞菌分泌的其它多糖也有一定降解能力。PslG在20-50攝氏度下均有活性,45度有最高多糖水解酶活。合成Psl胞外多糖的基因簇在假單胞菌中普遍存在,與此相一致的,PslG酶可瓦解多種假單胞菌形成的生物被膜。PslG酶不影 響細菌細胞的生長,對人上皮細胞無毒性。由于該PslG多糖水解酶可以迅速瓦解并分散假單胞菌形成的生物被膜,并能有效降解細菌分泌的胞外多糖,具有應用于驅油過程并提高原油采收率的巨大潛能。附圖說明圖1為經24h生長形成的銅綠假單胞菌氣液交界面生物被膜(亦被稱為菌膜)在PslG處理前和處理30分鐘后的激光共聚焦顯微鏡圖片。圖2為在Stovall三通道Flow-cell生物被膜培養系統中生長36h后形成的銅綠假單胞菌生物被膜經PslG處理后的激光共聚焦顯微鏡圖片。圖3為PslG多糖水解酶體外降解Psl多糖及其在不同溫度下的對Psl多糖的降解速率。其中,A圖所示為4mg/ml及2mg/mlPsl多糖與PslG于30℃共孵育1h前后的免疫點雜交結果,相應點跡計算后Psl多糖濃度標記于相應點上方;B圖顯示的是不同溫度下PslG降解Psl多糖的效率。圖4為不同假單胞菌中均有合成Psl多糖的同源基因簇。其中,gene表示基因。圖5為假單胞菌屬細菌的PslG具有很高的相似性。圖6為Psl多糖水解酶對產不同胞外多糖的銅綠假單胞菌菌株及其它假單胞菌形成的生物被膜均有瓦解作用。其中,A為Psl多糖水解酶對銅綠假單胞菌(MSH3,MSH10,CIM51,CIM52,CIM53)生物被膜的瓦解作用,B為Psl多糖水解酶對PAO1、PAO1△psl菌株、PAO1PBAD-psl菌株、粘性菌株PDO300、MJK8菌株、MJK8△psl菌株和MJK8△pel菌株生物被膜的瓦解作用,C為Psl多糖水解酶對斯氏假單胞菌A1501生物被膜的瓦解作用。圖7為PslG多糖水解酶對細菌生長的影響。圖8為PslG多糖水解酶對Caco2及HT-29的影響。具體實施方式下面結合具體實施方式對本發明進行進一步的詳細描述,給出的實施例僅為了闡明本發明,而不是為了限制本發明的范圍。下述實施例中的實驗方法,如無特殊說明,均為常規方法。下述實施例中所用的材料、試劑等,如無特殊說明,均可從商業途徑得到。下述實施例中的LBNS液體培養基的溶劑為水,其溶質及濃度分別為酵母粉5g/L,蛋白胨10g/L。LBNS固體平板為向LBNS液體培養基中加入瓊脂粉得到的固體培養基。下述實施例中的銅綠假單胞菌MSH3、MSH10、CIM51、CIM52、CIM53、PAO1、PAO1△psl菌株、PAO1PBAD-psl菌株、粘性菌株PDO300、MJK8菌株、MJK8△psl菌株和MJK8△pel菌株以斯氏假單胞菌A1501的菌株信息如下表所示:表、菌株信息上表中的文獻如下:Holloway,B.W.(1955).GeneticrecombinationinPseudomonasaeruginosa.JGenMicrobiol13,572-581.Ma,L.,Jackson,K.D.,Landry,R.M.,Parsek,M.R.,andWozniak,D.J.(2006).AnalysisofPseudomonasaeruginosaconditionalpslvariantsrevealsrolesforthepslpolysaccharideinadhesionandmaintainingbiofilmstructurepostattachment.JBacteriol188,8213-8221.Mathee,K.,Ciofu,O.,Sternberg,C.,Lindum,P.W.,Campbell,J.I.,Jensen,P.,Johnsen,A.H.,Givskov,M.,Ohman,D.E.,Molin,S.,etal.(1999).MucoidconversionofPseudomonasaeruginosabyhydrogenperoxide:amechanismforvirulenceactivationinthecysticfibrosislung.Microbiology145(Pt6), 1349-1357.Starkey,M.,Hickman,J.H.,Ma,L.,Zhang,N.,DeLong,S.,Hinz,A.,Palacios,S.,Manoil,C.,Kirisits,M.J.,Starner,T.D.,etal.(2009).Pseudomonasaeruginosarugosesmall-colonyvariantshaveadaptationsthatlikelypromotepersistenceinthecysticfibrosislung.JBacteriol191,3492-3503.Wolfgang,M.C.,Kulasekara,B.R.,Liang,X.,Boyd,D.,Wu,K.,Yang,Q.,Miyada,C.G.,andLory,S.(2003).ConservationofgenomecontentandvirulencedeterminantsamongclinicalandenvironmentalisolatesofPseudomonasaeruginosa.ProcNatlAcadSciUSA100,8484-8489YanY,PingS,PengJetal.Globaltranscriptionalanalysisofnitrogenfixationandammoniumrepressioninroot-associatedPseudomonasstutzeriA1501.BMCGenomics2010;11:11.下述實施例中的PslG多糖水解酶(PslG)的氨基酸序列如序列表中序列2所示,其編碼序列為序列1。按照如下方法制備:用序列1所示的DNA分子替換pET15b多克隆位點中兩個位點間的DNA片段,其他序列均不變,得到重組表達質粒PGL01-pslG。PGL01-pslG表達序列2所示的PslG多糖水解酶(PslG)。將表達質粒PGL01-pslG轉入大腸桿菌BL21(DE3),37度培養至OD0.8后加入0.12mM硫代半乳糖苷于22℃誘導過夜。4200rpm離心菌液15分鐘后重懸于結合緩沖液(25mMTris-HClpH8.0,200mMNaCl)中。將重懸液超聲破碎,12000rpm離心45分鐘去除沉淀。含有PslG蛋白的上清液過鎳親和柱并用結合緩沖液沖洗幾次去除非特異性結合的蛋白。結合在鎳柱樹脂上的蛋白用混有磷酸酯酶(終濃度為0.12mg/ml)的結合緩沖液重溶。將混合液在4℃孵育過夜以去除His標簽后將蛋白樣品用結合緩沖液洗脫。為進一步純化蛋白,將PslG過離子交換柱(Source15QHR16/10,GEHealthcare)并用0-1MNaClin25mMTris-HCl,pH8.0線性濃度梯度的緩沖液依次洗脫。最終,將蛋白樣品與含100mMNaCl的10mMTris-HClpH8.0緩沖液過分子篩(Superdex20010/300GL,GEHealthcare)后再用離子交換柱和分子篩純化一遍即得到PslG多糖水解酶。下述實施例中的Jensen培養基(JensenSE,FacyczIT,CampbellJN.NutritionalfactorscontrollingexocellularproteaseproductionbyPseudomonasaeruginosa.JBacteriol1980;144:844-847.)由溶質和溶劑組成,溶劑為水,溶質及其濃度分別為:NaCl5g/L,K2HPO43.286g/L,谷氨酸15.56g/L,纈氨酸2.81g/L,苯丙氨酸1.32g/L,葡萄糖12.81g/L,MgSO4·7H2O0.33g/L, CaCl2·2H2O0.021g/L,FeSO4·7H2O0.0011g/L,ZnSO4·7H2O0.0024g/L。下述實施例中的K培養基由溶質和溶劑組成,溶劑為水,溶質及其濃度分別為:K2HPO41.76g/L,KH2PO40.87g/L,MgSO4·7H2O0.29g/L,NaCl0.48g/L,乳酸鈉(60%儲液)6.3ml/L,CaCl2·2H2O70mg/L,FeCl3·6H2O10mg/L,NaMoO4·2H2O5mg/L,MnSO4·H2O250mg/L,ZnSO4·7H2O72mg/L,CuSO4·5H2O12.5mg/L,CoSO4·7H2O14mg/L,H3BO33mg/L。實施例1、PslG多糖水解酶可以降解Psl多糖從而瓦解分散銅綠假單胞菌生物被膜圖1所示為經24h生長形成的銅綠假單胞菌氣液交界面生物被膜(亦被稱為菌膜)在50nMPslG處理前和處理30分鐘后的激光共聚焦顯微鏡圖片。生物被膜和Psl多糖生物量及比例尺均已在相應圖片中標識。具體實施方法如下,實驗重復三次:從LBNS固體平板上挑新鮮銅綠假單胞菌PAO1菌株菌落于LBNS液體培養基中,在37攝氏度下200轉/分鐘震蕩培養過夜。然后以1%接種量接在含有1毫升Jensen基本培養基的4孔玻璃小腔室(Nunc,coverglasssystem,德國)中,在30攝氏度下靜止培養24小時以形成浮于培養液表面的菌膜。在已生長菌膜的腔室中添加100μl含PslG多糖水解酶并且PslG多糖水解酶濃度為50nM的Jensen培養液,于30攝氏度靜止培養30分鐘后,棄去游離的菌體,用熒光染料SYTO9標染生物被膜中的細菌細胞,熒光標記的凝集素HHA標染Psl胞外多糖,利用激光共聚焦熒光顯微鏡(OlympusFV1000,日本)對處理過后的菌膜進行觀測并用COMSTAT軟件進行數據分析,結果如圖1所示。按照上述方法,不加入PslG多糖水解酶,其他步驟均不變,得到未經PslG多糖水解酶處理的銅綠假單胞菌,作為對照,其結果如圖1所示。從圖1可以看出,經過PslG多糖水解酶處理半小時后的銅綠假單胞菌菌膜生物量為4.47±0.05μm3/μm2,Psl多糖生物量為2.54±0.09μm3/μm2;未經PslG多糖水解酶處理的銅綠假單胞菌菌膜生物量為1.61±0.83μm3/μm2,Psl多糖生物量為0.21±0.07μm3/μm2。經過PslG處理半小時后的銅綠假單胞菌菌膜較對照降低64%,而菌膜中的Psl胞外多糖降低了92%。由此說明,外源加入的PslG多糖水解酶可有效的降解Psl多糖,從而瓦解并分散生物被膜從而降低其生物量。實施例2、PslG對銅綠假單胞菌生物被膜的瓦解分散效率圖2所示為在Stovall公司三通道Flow-cell生物被膜培養系統中生長36h后形成的銅綠假單胞菌生物被膜經50nMPslG處理后的激光共聚焦顯微鏡圖片。相應的生物被膜平均厚度及生物量亦標于圖側。具體實施方法如下,實驗重復三次:用對數期銅綠假單胞菌PAO1接種入Stovall公司三通道Flow-cell,經1小時吸附后通Jensen培養基于室溫連續培養36h。生長36h的銅綠假單胞菌生物被膜用SYTO9標記后流入含有PslG并且50nMPslG多糖水解酶濃度為50nM的Jensen培養基,將PslG多糖水解酶流入時記為PslG處理0分鐘,在激光共聚焦顯微鏡(OlympusFV1000,日本)下PslG處理0分鐘開始對生物被膜連續觀察180分鐘,PslG處理0、30和180分鐘的結果如圖2所示。結果顯示,在PslG處理0分鐘,生物被膜的厚度為59.7±6.6μm,生物被膜生物量為47±5.3μm3/μm2;在PslG處理30分鐘,生物被膜的厚度為15.3±0.8μm,生物被膜生物量為19±1.0μm3/μm2;在PslG處理180分鐘,生物被膜的厚度為0.97±0.1μm,生物被膜生物量為0.72±0.1μm3/μm2。在PslG處理0-30分鐘內,PslG瓦解分散了44微米厚的生物被膜,每分鐘約驅散1.5微米生物被膜。之后的150分鐘中PslG仍保持活性,雖然效率略有下降。總之,經過180分鐘的PslG處理,99%的生物被膜生物量可被完全瓦解分散。實施例3、PslG多糖水解酶體外降解Psl多糖及其在不同溫度下的酶活圖3為PslG多糖水解酶體外降解Psl多糖及其在不同溫度下的對Psl多糖的降解速率。圖3中A圖所示為4mg/ml及2mg/mlPsl多糖與50nMPslG于30℃共孵育1h前后的免疫點雜交結果。相應點跡計算后Psl多糖濃度標記于相應點上方。圖3中B圖顯示的是不同溫度下50nMPslG降解Psl多糖的效率。具體實施方法如下,實驗重復三次:Psl多糖的制備和純化參照已發表文獻(ByrdMS,SadovskayaI,VinogradovEetal.GeneticandbiochemicalanalysesofthePseudomonasaeruginosaPslexopolysacchariderevealoverlappingrolesforpolysaccharidesynthesisenzymesinPslandLPSproduction.MolMicrobiol2009;73:622-638)進行。按照如下方法制備標準曲線:上述方法制備得到的Psl多糖進行10倍的梯度稀釋通過免疫印跡法檢測Psl多糖。向溶液1(PBS磷酸緩沖液)中加入Psl多糖與50nMPslG多糖水解酶,得到反應體系1,反應體系1中Psl多糖的濃度為4mg/ml,PslG多糖水解酶的濃度為50nM。將八個相同的反應體系1分別在20℃、25℃、30℃、37℃、40℃、45℃、50℃和55℃下孵育一小時后,用Psl多糖抗體(Psl多糖為以Psl多糖為免疫原得到的抗體)通過免疫印跡法檢測剩余的Psl多糖,通過QuantityOne(Bio-Rad)軟件進行分析處理后的信號強度并和標準曲線進行對比以計算PslG處理后Psl起始濃度為4mg/ml下的Psl的剩余量。按照上述方法,將Psl多糖的濃度為4mg/ml替換為Psl多糖的濃度為2mg/ml, 其他步驟均不變,得到PslG處理后Psl起始濃度為2mg/ml下的Psl的剩余量。按照上述方法,將50nMPslG多糖水解酶替換為0nMPslG多糖水解酶,得到PslG未處理的Psl起始濃度為4mg/ml下的Psl的剩余量。按照上述方法,將Psl多糖的濃度為4mg/ml替換為Psl多糖的濃度為2mg/ml,并將50nMPslG多糖水解酶替換為0nMPslG多糖水解酶,得到PslG未處理的Psl起始濃度為2mg/ml下的Psl的剩余量。50nMPslG與0nMPslG在30℃下處理Psl多糖1小時后后結果如圖3中A所示,結果顯示,經過PslG處理后的Psl多糖較未處理對照的量降低75%。由此說明,外源加入的PslG多糖水解酶能夠在體外實驗條件下,有效降解Psl多糖。計算PslG在不同溫度下對Psl多糖的降解速率(圖3中B),結果表明,PslG在45℃時的降解速率最高,在20-55℃下均有活性。實施例4、Psl多糖合成基因簇在假單胞菌屬的細菌中保守存在在基因數據庫GenBank與假單胞細菌研究網站(www.pseudomonas.com)中通過基因序列同源比對分析尋找與Psl多糖合成基因簇相似的基因簇,分析結果表明,在銅綠假單胞菌、熒光假單胞菌、惡臭假單胞菌、丁香假單胞菌等不同假單胞菌中均有合成Psl多糖的同源基因簇(圖4)。通過ESPript軟件,將來源于不同假單胞菌的PslG的氨基酸序列進行了比對,如圖5所示,來源于不同假單胞菌的PslG具有高度的同一性,特別是PslG多糖水解酶的保守活性結構域。實施例5、Psl多糖水解酶對產不同胞外多糖的銅綠假單胞菌菌株及其它假單胞菌形成的生物被膜均有瓦解作用為檢測PslG對不同來源菌株形成的生物被膜的作用,申請人使用了5株水環境來源的銅綠假單胞菌(MSH3,MSH10,CIM51,CIM52,CIM53),以及產生不同種胞外多糖的銅綠假單胞菌菌株:包括PAO1、只產生Pel胞外多糖的PAO1△psl菌株,過表達產生大量Psl胞外多糖的PAO1PBAD-psl菌株,產生褐藻多糖的粘性菌株PDO300,過量產生Psl與Pel胞外多糖的MJK8菌株,以及只過量產生Pel胞外多糖的MJK8△psl菌株和只過量產生Psl胞外多糖的MJK8△pel菌株。此外,還檢測了PslG對斯氏假單胞菌A1501生物被膜的作用。具體實施方法如下,實驗重復三次:銅綠假單胞菌各菌株按照如下方法操作:接LBNS平板上新鮮單克隆于LBNS液體培養基中,在37度下200轉/分鐘震蕩培養12小時。然后以1%接種量接在Jensen培養液中,于96孔板中30度靜止培養24小時后,棄去游離的菌體,用0.8%生理鹽水洗3次后添加100μl含有PslG的Jensen培養液(含有PslG的Jensen培養液中PslG的濃度為50nM),不同時間處理后,用結晶紫染色法檢測處理后的生物被膜的生物量。 同時以未加PslG的Jensen培養液為對照組。生物被膜瓦解比例為處理組與對照組相比減少的生物量除以對照組生物量的百分比。斯氏假單胞菌按照如下方法操作:接過夜培養的斯氏假單胞菌菌液于含50nMPslG的K氏培養基(含有PslG的K氏培養基中PslG的濃度為50nM)中,于30℃培養48h后用體視顯微鏡觀察菌膜的形成情況,同時以未加PslG的K氏培養基為對照組。相應生物被膜的生物量亦用結晶紫染色法檢測,并在OD560下進行檢測定量。結果如圖6中A和B所示,PslG對產不同胞外多糖的銅綠假單胞菌菌株形成的生物被膜均有一定瓦解作用。說明PslG對其它多糖(如Pel多糖,褐藻多糖等)也有一定降解活性。表明,PslG對不同來源的多株銅綠假單胞菌的生物被膜具有抑制效果。PslG對斯氏假單胞菌的生物被膜也有一定瓦解作用,未經PslG處理的斯氏假單胞菌的生物被膜生物量在OD560下的檢測結果為10.1±0.8,PslG處理的斯氏假單胞菌的生物被膜生物量在OD560下的檢測結果為2.7±0.8,極顯著低于未經PslG處理的斯氏假單胞菌的生物被膜生物量。說明PslG有一定的廣譜活性,適用范圍較廣。實施例6、PslG多糖水解酶對細胞無毒性,不影響細菌的生長實驗重復三次,具體實施方法如下:1、PslG對銅綠假單胞菌生長的影響于含不同濃度PslG(0nM,1nM,50nM,100nM)的Jensen培養基中1%接種過夜的銅綠假單胞菌PAO1菌液,于37℃振蕩培養24h并連續取樣測定OD600值。結果如圖7所示,表明,外源添加不同濃度PslG對綠假單胞菌菌株PAO1的生長無影響,說明PslG多糖水解酶對細菌細胞的生長無影響,表明PslG多糖水解酶對細菌細胞無毒性。2、PslG對人的細胞的影響實驗重復三次,實驗方法如下:以104個細胞/孔置人腸道上皮細胞系Caco2于96孔板中培養12小時,再繼續用含不同濃度PslG蛋白的DMEM培養基(PslG蛋白的的濃度分別為0、50和100nM)繼續培養12小時后,每孔加入10μl5mg/mlMTT,在37℃培養4小時。生成的有色甲晶體用100μlDMSO溶解后讀取OD540(圖8)。按照上述方法,將Caco2替換為HT-29,其他步驟均不變,檢測PslG對HT-29的影響(圖8)。結果顯示:高濃度PslG蛋白對人上皮細胞無毒性。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1