一種光敏聚酰亞胺及其制備方法和應用與流程
本發明屬于高分子材料制備,更具體地說,本發明涉及一種光敏聚酰亞胺及其制備方法和應用。
背景技術:
1、聚酰亞胺是一種性能高超的高分子材料,通常是指主鏈中含有酰亞胺環的一類聚合物。因其具有優異的熱穩定性、機械性能、耐化學性、良好的介電常數和優異的成像性能,被廣泛應用于微電子、航空航天、石油化工、特種防護和光電材料等多個領域。聚酰亞胺樹脂作為耐熱等級最高的樹脂之一,現有報道中,長期使用現有報道中的長期使用溫度可達400℃,短期使用甚至可以達到550℃。聚酰亞胺分子具有很高的可設計性,可以通過調整主鏈二胺和二酐的種類、比例,也可以通過側鏈改性、合成后封端、納米雜化等手段調整分子結構,得到滿足特定要求的聚酰亞胺材料。隨著工業化進程的不斷加快,對聚酰亞胺材料的性能和微加工技術提出了更深層次的要求。聚酰亞胺的傳統制造工藝包括熱成型、熱壓灌注成型和冷壓燒結成型。上述工藝需要高溫、高壓、模具輔助等苛刻條件,存在高能耗、高成本、危險性、復雜性等問題,難以進行高自由度設計和微尺寸精細加工。因此,近年來許多學者嘗試將聚酰亞胺材料應用到新的快速成型制造技術中,以解決上述問題,實現精密加工的技術需求。
2、數字光處理式3d打印技術(dlp技術),是將影像信號經過數字處理后,再通過投影儀將光投射到液態光敏樹脂表面,層層固化成型的增材制造技術。相比于其他3d打印方式,數字光處理具有精度高(精加工尺度可達20~30μm)、表面光滑、打印成型速度快、節約成本、無活動部件,振動偏差小等優點,作為一種高效,精確,高質量的快速成型技術,已經在多個領域取得了廣泛的應用。因而,開發一種高溶解性的,具有光敏活性的,可以用于數字光處理式3d打印技術的聚酰亞胺樹脂,對于解決聚酰亞胺難以成型,無法制備精密結構的問題,具有十分深遠乃至革命性的意義。
3、應用于數字光處理式3d打印技術的樹脂,通常來說需要具有較好的光敏活性,無溶劑的流動性或在溶劑中較高的溶解度,黏性較小,無毒,不易在空氣中發生反應等性能。常規聚酰亞胺材料由于其中剛性較強的單體使得分子鏈和較強的分子間作用力,導致其玻璃化轉變溫度高、熔點高、熔體黏度高,同時大多常規結構的聚酰亞胺樹脂在有機溶劑中溶解性較差,常規加工方法同樣不易實現,更難應用于數字光處理式3d打印技術。此外,3d打印技術所需要的樹脂漿料需要盡可能提高固體含量,這是由于后固化過程中,溶劑隨固化揮發,體積會相應地產生坍縮,破壞原本的結構。聚合物溶解過程分為溶脹和溶解兩個階段。在溶脹階段,溶劑分子向聚合物擴散,導致大分子體積膨脹;在溶解階段,在溶劑分子的作用下,大分子之間的作用力不斷減弱,最終分散到溶劑中,與溶劑分子相互混合,形成分子分散的均相體系。因而,增加溶解度的關鍵,即是降低溶劑小分子向聚合物內擴散的難度,降低聚合物鏈之間的作用力,以及增加聚合物與溶劑小分子之間的作用力,使其更容易分散。
技術實現思路
1、本發明的一個目的是解決至少上述問題和/或缺陷,并提供至少后面將說明的優點。
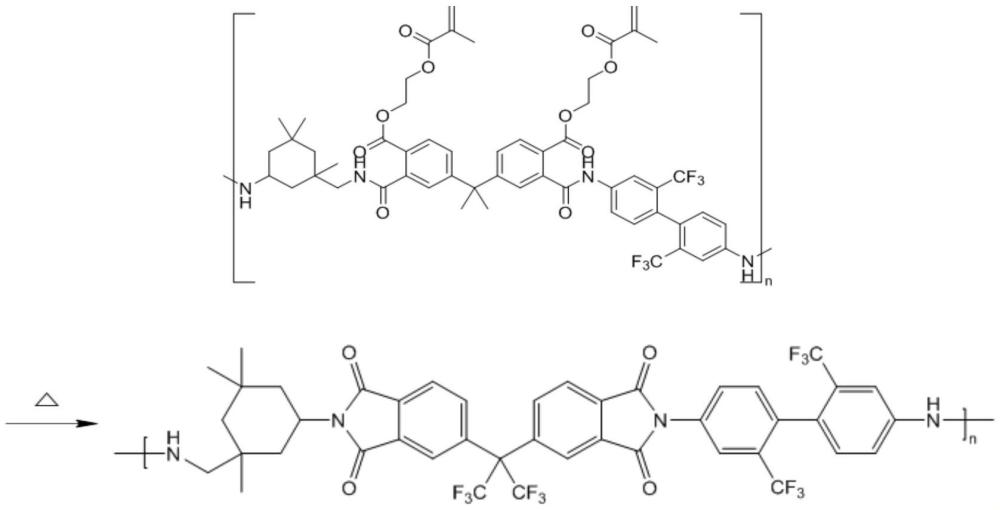
2、為了實現本發明的這些目的和其它優點,提供了一種光敏聚酰亞胺,其化學結構式為:
3、其中,n=4~6。
4、一種如上所述的光敏聚酰亞胺的制備方法,包括以下步驟:
5、步驟一、在無水無氧條件下,將4,4'-(六氟異丙烯)二酞酸酐溶于n-甲基吡咯烷酮中,置于反應器內,將異佛爾酮二胺和2,2'-雙(三氟甲基)二氨基聯苯溶于n-甲基吡咯烷酮中得到二胺溶液,將二胺溶液逐滴加入反應器內,保持攪拌,反應6~10h,得到黏稠的聚合物溶液;
6、步驟二、向反應器中加入n-甲基吡咯烷酮稀釋聚合物溶液,將n,n'-二環己基碳二亞胺溶于n-甲基吡咯烷酮中并逐滴加入反應器內,保持攪拌,反應2~4h;
7、步驟三、將甲基丙烯酸羥乙酯和n,n-二甲基芐胺溶于n-甲基吡咯烷酮中,關閉室內其他光源,以黃色燈光作為可視光源,遮蔽反應器,將前述溶液逐滴加入反應器內,并加熱到45~60℃,保持攪拌,反應15~24h,得到黏稠淡黃色溶液;
8、步驟四、將得到的黏稠淡黃色溶液加入布氏抽濾漏斗中,抽濾,收集濾液;
9、步驟五、將濾液加入到以400~800rpm轉速攪拌的甲醇中,封閉反應器,在避光條件下繼續攪拌1~3h,置換樹脂中殘留的高沸點溶劑;
10、步驟六、將步驟五得到的產物置于布氏抽濾漏斗中,抽濾,收集沉淀物,置于放有五氧化二磷的真空干燥箱中,在真空度-1~-0.3bar、25~40℃條件下干燥10~16h;
11、步驟七、收集干燥后的產物加入到n-甲基吡咯烷酮中,攪拌溶解,重復步驟五和步驟六,直到得到泡沫狀的疏松白色聚合物,即為光敏聚酰亞胺的前驅體聚酰胺酸;
12、步驟八、前驅體聚酰胺酸經熱處理,得到光敏聚酰亞胺;或將前驅體聚酰胺酸與有機溶劑混合,再加入光引發劑和活性稀釋劑,加熱至70~80℃,攪拌至完全溶解,得到聚酰胺酸溶液,然后通過澆鑄、刮涂或3d打印的成型方法,經干燥、光固化和熱處理,得到光敏聚酰亞胺的材料。
13、優選的是,所述步驟一中,4,4'-(六氟異丙烯)二酞酸酐、異佛爾酮二胺和2,2'-雙(三氟甲基)二氨基聯苯的摩爾比為2:0.5~1.5:0.5~1.5;4,4'-(六氟異丙烯)二酞酸酐和n-甲基吡咯烷酮的摩爾體積比為1mol:1~2l;異佛爾酮二胺和2,2'-雙(三氟甲基)二氨基聯苯總摩爾與n-甲基吡咯烷酮體積的比例為1mol:300~500ml;逐滴加入速度為15~25滴/min。
14、優選的是,所述步驟二中,向反應器中加入的n-甲基吡咯烷酮與步驟一中的4,4'-(六氟異丙烯)二酞酸酐的體積摩爾比為200~400ml:1mol;n,n'-二環己基碳二亞胺和步驟一中的4,4'-(六氟異丙烯)二酞酸酐的摩爾比為2~6:1;n,n'-二環己基碳二亞胺和n-甲基吡咯烷酮的摩爾體積比為2~6mol:1~2l;逐滴加入速度為15~25滴/min。
15、優選的是,所述步驟三中,甲基丙烯酸羥乙酯和步驟一中的4,4'-(六氟異丙烯)二酞酸酐的摩爾比為0.1~6:1;甲基丙烯酸羥乙酯、n,n-二甲基芐胺和n-甲基吡咯烷酮的摩爾體積比為0.1~6mol:0.02~0.2mol:200~400ml;黃色燈光為20~40w;逐滴加入速度為15~25滴/min。
16、優選的是,所述步驟五中,甲醇與步驟一中的4,4'-(六氟異丙烯)二酞酸酐的體積摩爾比為5~8l:2mol;所述步驟七中,n-甲基吡咯烷酮與步驟一中的4,4'-(六氟異丙烯)二酞酸酐的體積摩爾比為1~2l:1mol。
17、優選的是,所述步驟八中,前驅體聚酰胺酸和有機溶劑的質量比為1:0.5~1.5;以前驅體聚酰胺酸的質量為100%計,光引發劑的添加量為1~5%質量分數,活性稀釋劑的添加量為1~15%質量分數。
18、優選的是,所述步驟八中,有機溶劑為n-甲基吡咯烷酮(nmp)、n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dmac)、四氫呋喃(thf)、1,4-二氧六環、二甲基亞砜(dmso)、n-乙烯基吡咯烷酮(nvp)中的一種或多種;光引發劑為2,4,6-三甲基苯甲酰基苯基膦酸乙酯(tpo-l)、苯基雙(2,4,6-三甲基苯甲酰基)氧化膦(光引發劑819)、4,4'-雙(二甲基氨基)二苯甲酮(deak)、1-羥環己基苯酮(光引發劑184)中的一種或多種;活性稀釋劑為三羥甲基丙烷三丙烯酸酯(tmpta)、1,6-己二醇二丙烯酸酯(hdda)、季戊四醇三丙烯酸酯(peta)中的一種或多種。
19、優選的是,所述步驟八中,刮涂成型的具體方法為:將聚酰胺酸溶液均勻涂布于基材上,置于真空干燥箱中,在真空度-0.6~-0.1bar、70~90℃條件下干燥6~10h,得到聚酰胺酸薄膜,將聚酰胺酸薄膜置于60~80mw/cm2紫外燈下固化1~3min,然后在氬氣氣氛中,以0.5~5℃/min的升溫速率升溫至200~230℃,保溫0.5~2h,自然冷卻,得到光敏聚酰亞胺薄膜;所述基材包括玻璃板、石英板、硅片或金屬片。
20、優選的是,所述步驟八中,澆鑄成型的具體方法為:將聚酰胺酸溶液轉移到帶有魯爾接頭的點膠針管內,均勻擠出澆鑄在給定結構的模具中,置于真空干燥箱中,在真空度-0.6~-0.1bar、70~90℃條件下干燥6~10h,得到聚酰胺酸塊體,將聚酰胺酸塊體置于60~110mw/cm2紫外燈下固化3~10min,然后在氬氣氣氛中,以0.5~5℃/min的升溫速率升溫至200~230℃,保溫0.5~2h,自然冷卻,得到光敏聚酰亞胺塊體。
21、優選的是,所述步驟八中,3d打印成型包括直寫式3d打印和數字光處理式(dlp)3d打印。
22、優選的是,所述直寫式3d打印為:將固含量40%以上的聚酰胺酸溶液轉移到帶有螺紋接口的點膠針管中并接上打印針頭,連接直寫式打印裝置,以壓縮氣體作為動力源,調節氣體壓力為100~800kpa,按照預先設置的模型在基底上開始直寫式打印,同時以60~110mw/cm2的紫外光輔助固化成型,打印完成后,將模型置于60~110mw/cm2紫外燈下后固化3~10min,然后在氬氣氣氛中,以0.5~5℃/min的升溫速率升溫至200~230℃,保溫0.5~2h,自然冷卻,得到光敏聚酰亞胺成品;所述壓縮氣體包括壓縮空氣、氬氣或氮氣,其供氣形式為空壓機或壓力氣罐;所述基底包括玻璃板、石英板、硅片或金屬片。
23、優選的是,所述數字光處理式3d打印為:將聚酰胺酸溶液倒入裝有離型膜的dlp打印機樹脂槽內,設置打印參數,開始打印,打印完成后用刀片從打印臺上取下模型,用潔凈的乙醇沖洗,然后將模型置于60~110mw/cm2紫外燈下后固化3~10min,然后在氬氣氣氛中,以0.5~5℃/min的升溫速率升溫至200~230℃,保溫0.5~2h,自然冷卻,得到光敏聚酰亞胺成品;所述打印參數為:底層曝光時間28~40s,更優選為35s,底層層數1~20層,更優選為8層,層厚度0.1mm~0.05mm,每層固化時間2~10s。
24、優選的是,所述步驟三中,甲基丙烯酸羥乙酯和步驟一中的4,4'-(六氟異丙烯)二酞酸酐的摩爾比<1時,加入甲醇封閉反應,即所述步驟三替換為:將甲基丙烯酸羥乙酯和n,n-二甲基芐胺溶于n-甲基吡咯烷酮中,關閉室內其他光源,以黃色燈光作為可視光源,遮蔽反應器,將前述溶液逐滴加入反應器內,并加熱到45~60℃,保持攪拌,反應10~12h,然后將甲醇和n,n-二甲基芐胺溶于n-甲基吡咯烷酮中,逐滴加入反應器內,并加熱到45~60℃,保持攪拌,反應10~12h,得到黏稠淡黃色溶液。
25、優選的是,所述甲基丙烯酸羥乙酯和甲醇的摩爾比為0.1~2:3~4;甲醇、n,n-二甲基芐胺和n-甲基吡咯烷酮的摩爾體積比為3~4mol:0.02~0.2mol:100~300ml。
26、優選的是,所述步驟一到步驟三替換為:
27、步驟一、在充有氬氣保護的手套箱中(氧含量≤0.5ppm,水含量≤0.5ppm),放置試管型高速剪切分散機,并置入可替換的分散管;在手套箱內,將4,4'-(六氟異丙烯)二酞酸酐置于分散管內,滴加無水級n-甲基吡咯烷酮,關閉分散管,7000~9000rpm攪拌分散;將異佛爾酮二胺和2,2'-雙(三氟甲基)二氨基聯苯溶于無水級n-甲基吡咯烷酮中得到二胺溶液,每隔1min開啟分散管,滴入2滴二胺溶液,直到二胺溶液全部滴加完畢,保持7000~9000rpm攪拌,反應6~10h,得到黏稠的聚合物溶液,用椎板粘度計檢測黏度,當黏度達到8000cp,即可進行步驟二;
28、步驟二、將n,n'-二環己基碳二亞胺溶于n-甲基吡咯烷酮中并采用步驟一相同的方法滴加入分散管內,保持攪拌,反應2~4h;
29、步驟三、將甲基丙烯酸羥乙酯和n,n-二甲基芐胺溶于n-甲基吡咯烷酮中,關閉室內其他光源和手套箱內白色光源,以黃色燈光作為可視光源,并采用步驟一相同的方法滴加入分散管內,并加熱到45~60℃,保持攪拌,反應15~24h,得到黏稠淡黃色溶液。
30、一種如上所述的制備方法制備的光敏聚酰亞胺在3d打印中的應用。
31、一種如上所述的制備方法制備的光敏聚酰亞胺的前驅體聚酰胺酸在3d打印中的應用。
32、本發明至少包括以下有益效果:針對現有聚酰亞胺樹脂不具有光敏活性、難以熔融和溶解、無法應用于3d打印技術、部分聚酰亞胺材料有高毒性等問題,本發明提供了一種低分子量、引入大位阻柔性基團、接枝光敏酯基側鏈的聚酰亞胺;
33、本發明將異佛爾酮二胺結構引入聚酰亞胺的結構中,使分子鏈產生折疊和扭轉,充分增強了其溶解性能,通過調節異佛爾酮二胺和2,2'-雙(三氟甲基)二氨基聯苯的比例調節樹脂的溶解性、力學性能和分子量,同時采用n,n'-二環己基碳二亞胺作為關環縮合劑,使樹脂先形成聚異酰亞胺結構中間體,再利用含有醇羥基的光敏單體甲基丙烯酸羥乙酯在n,n-二甲基芐胺的催化下開環,最終得到具有光敏活性的聚酰亞胺前驅體聚酰胺酸,可以通過調節甲基丙烯酸羥乙酯和甲醇的比例調節樹脂的光敏活性,適應不同場景下對光敏樹脂活性的要求;
34、本發明所制備的前驅體聚酰胺酸具有較強的光敏活性,以及較好的溶解性能,在n,n-二甲基甲酰胺中溶解度可以達到56wt%,可用于光固化工藝和數字光處理式3d打印,在經熱處理后可以得到力學性能較好的聚酰亞胺,并且制備得到的聚酰亞胺毒性較小,透明度較高,耐高溫,具有光刻等領域的應用前景;
35、此外,由于酸酐極易水解變質,氧也會干擾有機合成反應,故而合成聚酰亞胺樹脂需要無水無氧、氬氣或氮氣保護的環境;使用傳統的機械攪拌的合成方式,需要密封反應器,但仍然會密封不夠嚴實,通常解決的辦法是持續通入氬氣來保證惰性氣氛環境,而本發明采用高速剪切分散機,即不需要機械攪拌的較笨重的設備,也不需要密封,可以達到更好的無水無氧環境,反應得到的樹脂性能更好;此外,一旦反應物分子量提高溶液黏度會相應升高,導致攪拌困難,而本發明采用高速剪切分散機扭矩大,即使分子量高也能充分攪拌,可以使反應更完全、副反應和副產物更少,產率相對更高,得到的產物更穩定。
36、本發明的其它優點、目標和特征將部分通過下面的說明體現,部分還將通過對本發明的研究和實踐而為本領域的技術人員所理解。
- 還沒有人留言評論。精彩留言會獲得點贊!