甲酸脫氫酶工程菌株的構建方法及應用與流程
本發明涉及甲酸脫氫酶工程菌株的構建方法及應用,屬于基因工程技術領域。
背景技術:
氧化還原酶催化底物的氧化或還原,在生物體利用有機物的過程中起著至關重要的作用。氧化還原反應伴隨著電子的傳遞,生物體內絕大多數的氧化還原酶在反應時需要輔酶作為電子傳遞體。反應過程中,伴隨著有機物的氧化,還原態的nadh會被氧化為其氧化態形式nad+,當nadh被消耗殆盡,會導致反應的終止。生物體內有nad+<=>nadh的相互轉化系統可以持續為氧化還原酶提供還原態nadh,當氧化還原酶細胞外應用時,應為其持續提供還原態輔酶nadh,而甲酸脫氫酶(formatedehydrogenase,fdh)是最成功的輔酶再生體系之一,已應用于工業生產中。
中國專利文獻cn104561052a(申請號201410803992.1)公開了一種重組甲酸脫氫酶及其制備方法和應用。一種甲酸脫氫酶基因突變體,核苷酸序列如seqidno.1所示。一種重組甲酸脫氫酶,選自(1)由seqidno.2所示氨基酸序列組成的蛋白質;或是(2)由seqidno.2中所示的氨基酸序列經過取代、缺失或者添加一個或者幾個氨基酸且具有甲酸脫氫酶活性的由(1)衍生的蛋白質。一種用于生產重組甲酸脫氫酶的基因工程菌,并且通過培養該基因工程菌和優化發酵工藝,實現重組甲酸脫氫酶的工業化生產,將其用于輔酶nad+和nadh再生循環體系,并用于相關原料藥及醫藥中間體的生產。
中國專利文獻cn104087522a(申請號201410306082.2)公開了一種外源表達甲酸脫氫酶的酵母工程菌,其分類命名為巴斯德畢赤酵母(pichia.pastoris),已保藏于中國普通微生物菌種保藏管理中心,登記入冊編號為cgmccno:8924,保藏日期為2014年3月17日。本發明還公開了上述酵母工程菌的構建方法和應用。該發明的酵母工程菌經甲醇誘導后,測的粗酶活為27.9u/ml。
雖然上述工程菌均能夠表達甲酸脫氫酶,但受限于菌株的生長及酶活的原因,無法滿足市場的需求。
技術實現要素:
本發明針對現有技術的不足,提供甲酸脫氫酶工程菌株的構建方法及應用。
本發明技術方案如下:
甲酸脫氫酶工程菌株的構建方法,步驟如下:
(1)提取博伊丁假絲酵母基因組dna為模板;
(2)以步驟(1)制得的基因組dna為模板,以兼并引物進行pcr擴增,兼并引物的上游引物核苷酸序列如seqidno.1所示,兼并引物的下游引物核苷酸序列如seqidno.2所示,制得fdh基因片段;
(3)將步驟(2)制得的fdh基因片段用內切酶xbai和bamhi雙酶切fdh基因和質粒pet28a(+),再用連接酶連接,得到質粒pet28a(+)-fdh;
(4)將質粒pet28a(+)-fdh轉化進大腸桿菌bl21(de3)感受態細胞,涂布篩選平板,37℃培養24h后進行克隆子鑒定,選取陽性菌株,得到表達fdh的重組菌株bl21(de3)-fdh。
根據本發明優選的,所述步驟(1)中,博伊丁假絲酵母購自中國普通微生物菌種包藏管理中心(cgmcc),編號為:2.2378。
根據本發明優選的,所述步驟(2)中,pcr擴增條件為:94℃預變性3min;94℃30s,58℃30s,72℃45s,30循環;72℃延伸7min。
根據本發明優選的,所述步驟(2)中,pcr擴增體系如下,總體系50μl:
dntp5μl,上游引物1μl,上游引物1μl;模板dna1μl,taqdna聚合酶2.5u,buffer5μl,加雙蒸水至50μl。
根據本發明優選的,所述步驟(4)中,篩選平板組分如下,均為質量百分比:
1%蛋白胨,1%nacl,0.5%酵母提取物,2%瓊脂,100μg/ml卡那霉素,余量水。
上述甲酸脫氫酶工程菌株在多環芳烴降解過程中的應用。
有益效果
1、本發明中所述的引物對是根據ncbi中所登錄的多株博伊丁假絲酵母的fdh序列設計的兼并引物,可以以多數博伊丁假絲酵母菌株的基因組dna為模板擴增出fdh基因,而不受基因序列中個別堿基差異的影響。
2、利用本發明的菌株bl21(de3)-fdh制備的甲酸脫氫酶粗酶液酶活性高,可達到0.6331u/ml,具有很好的工業應用性。
附圖說明
圖1是從對菌株bl21(de3)-fdh中提取的質粒pet28a(+)-fdh進行酶切鑒定的瓊脂糖凝膠電泳圖;
其中:m泳道:dnamarker,1泳道:質粒pet28a(+)-fdh單酶切片段,2泳道:質粒pet28a(+)-fdh雙酶切片段;
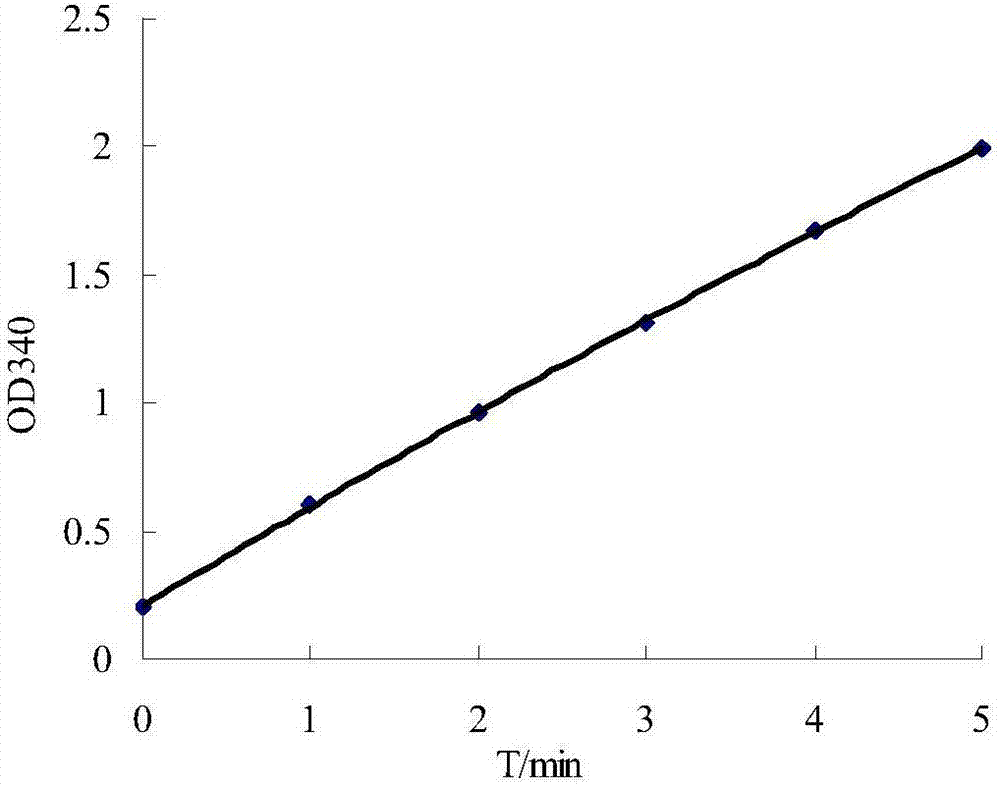
圖2是甲酸脫氫酶活性測定時吸光度od340隨時間的變化曲線;
圖3是菲降解率和降解時間的變化曲線。
具體實施方式
下面結合說明書及實施例對本發明的技術方案做進一步闡述,但本發明所保護范圍不限于此。
生物材料來源
實施例中所述博伊丁假絲酵母(candidaboidinii)購自中國普通微生物菌種包藏管理中心(cgmcc),菌種編號為:2.2378。
分子生物學試劑購自大連寶生物生物工程有限公司。
實施例1
菌株bl21(de3)-fdh的構建
提取博伊丁假絲酵母的基因組作為模板,用兼并引物對為引物進行pcr擴增,制得fdh基因片段;
上游兼并引物核苷酸序列如seqidno.1所示,下游兼并引物核苷酸序列如seqidno.2所示:
seqidno.1:5’-cctctagaaaggagatataatgaagatygtyttagtyytwtatg-3’
seqidno.2:5’-ccggatccttatttcttatcgtgtttaccg-3’
擴增條件為:94℃預變性3min;94℃30s,58℃30s,72℃45s,30循環;最后72℃延伸7min。
得到的fdh基因片段經內切酶xbai和bamhi雙酶切,與同樣經xbai和bamhi雙酶切的質粒pet28a(+)進行連接(連接產物為質粒pet28a(+)-fdh),連接8h后轉化bl21(de3)感受態,涂布含卡納霉素的固體lb平板進行,然后對平板上生長出的菌落進行挑篩選鑒定,酶切結果見圖1,命名為bl21(de3)-fdh。
篩選平板組分如下,均為質量百分比:
1%蛋白胨,1%nacl,0.5%酵母提取物,2%瓊脂,100μg/ml卡那霉素,余量水。
實施例2
菌株bl21(de3)-fdh甲酸脫氫酶活性的誘導表達及粗酶液制備
將bl21(de3)-fdh接種到5ml的lb液體培養基中,向培養基中加入5μl,100mg/ml的卡那霉素溶液,搖床中37℃、180r/min條件下進行培養12~24h。。
轉接3ml菌液至50ml新鮮的lb液體培養基中,在溫度為37℃,搖床轉速為180r/min條件下繼續培養,2~3h后,添加iptg溶液至終濃度0.5mmol/l進行誘導,誘導時間為3~5h。
離心收集菌體完全,用20~30ml的pbs緩沖液(0.1mph7.0)進行菌體重懸,然后超聲破碎(功率200~400w,條件為超聲3s,間歇3s,共10min),離心,上清即為粗酶液。
實施例3
菌株bl21(de3)-fdh甲酸脫氫酶活性的測定
酶活測定體系:反應液為0.2mlnad(16.7mm)、0.2ml甲酸鈉(1670mm)和1.4mlpbs緩沖液(ph7.5、10mm、含4mmdtt),加入0.2ml經稀釋過的粗酶液,混合后立即置于分光光度計,測340nm處的吸光值a1,室溫(20℃)反應5min,測340nm處吸光值a2,a2-a1用于計算酶活性,吸光值隨時間的變化曲線如圖2所示。
酶活力定義為:在ph7.5、30℃條件下,每分鐘生成1μmolnadh所需要的酶量為1個酶活單位(nadh的摩爾消光系數為6220l·mol-1·cm-1)。
經計算,酶活為0.6331u/ml。
實施例4
本實施例主要用于說明本發明制備的甲酸脫氫酶在多環芳烴降解過程中的應用及有益效果。
本實驗室前期篩選的多環芳烴降解菌多食鞘氨醇桿菌(sphingobacteriummultivorum),用lb培養基在搖床中32℃、150rpm條件下培養16h,利用超聲破碎儀在功率320w,超聲5s,間歇5s,共15min的條件下破碎,離心,取上清液,即為多環芳烴降解粗酶液。
將本實施例中的多環芳烴降解粗酶液與實施例2中制備的甲酸脫氫酶粗酶液混合,進行多環芳烴的降解。
體系為:反應總體積5ml,菲終濃度50mg/l,用pbs緩沖液(ph7.5、10mm、含4mmdtt)配制,0.5ml多環芳烴降解粗酶液以及0.5ml甲酸脫氫酶粗酶液;
對照體系為反應總體積5ml,菲終濃度50mg/l,用pbs緩沖液(ph7.5、10mm、含4mmdtt)配制,加入0.5ml多環芳烴降解粗酶液。
結果如圖3所示,由于nadh的消耗,反應3h后體系中的菲不再減少。
而添加甲酸脫氫酶的處理組中,由于實現了nadh的循環再生,因此一直到6h后,菲的降解都保持著很高的速度。
sequencelisting
<110>山東省科學院生態研究所
<120>甲酸脫氫酶工程菌株的構建方法及應用
<160>2
<170>patentinversion3.5
<210>1
<211>44
<212>dna
<213>人工合成
<400>1
cctctagaaaggagatataatgaagatygtyttagtyytwtatg44
<210>2
<211>30
<212>dna
<213>人工合成
<400>2
ccggatccttatttcttatcgtgtttaccg30
- 還沒有人留言評論。精彩留言會獲得點贊!