一種四元羧酸N,N,N′,N′?四(4′?羧基聯苯基)?1,4?苯二胺的合成方法與流程
本發明涉及有機化學的技術領域,尤其一種四元羧酸n,n,n′,n′-四(4′-羧基聯苯基)-1,4-苯二胺的合成方法。
背景技術:
延長的剛性四羧酸類配體廣泛應用于mofs材料的合成中,這類材料通常具有良好的孔道結構、大的比表面積,使這類材料在氣體分離與儲藏、化學傳感、催化及藥物緩釋等不同領域具有潛在應用。由于芳香四羧酸特別是長鏈的芳香四羧酸具有以下特性而成為制備高維金屬有機框架的較好選擇:一、可全部或部分去質子化的羧基可使該配體具有更為靈活多樣的配位模式;二、兩個鄰位的羧基距離較近,易于形成分子內氫鍵;三、由于自身大的空間位阻,苯環與苯環之間有一定的夾角,部分羧基也會和苯環有一定的夾角,可以從不同方向與金屬離子配位;四、分子較高的對稱性有利于晶體的生長與結構的設計。比如2009年lin(mal.,jina.,xiez.,etal.angew.chem.int.ed.,2009,48:9905-9908)等使用兩個四羧酸配體和銅鹽在溶劑熱條件下成功合成兩個金屬有機框架,作者使用冷凍干燥法有效增加了化合物的比表面積,同時也增加了氫氣的吸附量。使用冷凍干燥法比真空干燥的多一倍的氫吸附量。stylianou等(stylianouk.c.,heckr.,chongs.y.,etal.j.am.chem.soc.,2010,132:4119-4130)報道使用基于芘的四羧酸配體(1,3,6,8-四(4-苯甲酸)芘(tbapy))和硝酸銦合成一個強熒光的金屬有機框架1,1展現了良好的熱穩定性。實驗表明1在471和529nm處有強的熒光。化合物1的熒光壽命達到0.110ms,量子產率為6.7%。延長的剛性四角羧酸配體通常能得到結構新穎、比表面積大的多微孔mofs材料,并能改變微孔結構,使其微孔內電子環境得到優化從而提高上述應用性能,因此,設計合成結構新穎的延長多羧酸配體并應用于mofs材料的合成已成為當今研究的熱點之一。然而,大尺寸的剛性多羧酸化合物合成比較困難。因此本發明中所涉及的結構新穎的n,n,n′,n′-四(4′-羧基聯苯基)-1,4-苯二胺是一種具有巨大潛在應用價值的配體。目前尚無文獻報道該化合物。
技術實現要素:
本發明的目的是提供一種合成成本低、產率高、產品純度高的新型n,n,n′,n′-四(4′-羧基聯苯基)-1,4-苯二胺的合成方法。
本發明的n,n,n′,n′-四(4′-羧基聯苯基)-1,4-苯二胺的合成方法,合成步驟為:
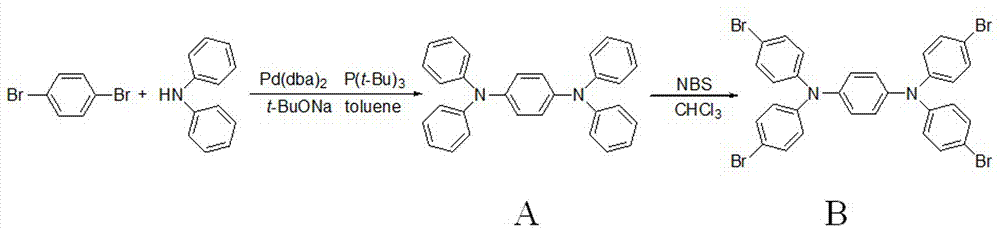
(ⅰ)以1,4-二溴苯和二苯胺為原料,加入一定量叔丁醇鈉和適量催化劑,加入甲苯作為溶劑,在氮氣或氬氣保護下快速加入三-(2-甲苯基)膦,摩爾比1,4-二溴苯:二苯胺:叔丁醇鈉為1:2.1~2.3:2.1~2.5,1,4-二溴苯與甲苯的用量比是1毫摩:3.0~4.5毫升。加完后在85~100℃油浴加熱反應10~16小時,反應結束蒸出甲苯,加20~40ml水稀釋,二氯甲烷萃取(20~40ml×3),收集有機相并用無水亞硫酸鈉對其進行除水處理,過濾,真空濃縮,最后以石油醚和乙酸乙酯按體積比為20:0.8~1.2的混合溶劑為洗脫劑用柱色譜法分離提純得到化合物a:n,n,n′,n′-四苯基-1,4-苯二胺;
(ⅱ)n,n,n′,n′-四苯基-1,4-苯二胺和nbs(n-溴代丁二酰亞胺)按摩爾比1:3.5~5.5混合后溶于一定溶劑中,n,n,n′,n′-四苯基-1,4-苯二胺與溶劑的用量比是1毫摩:6.0~8.0毫升,在0~30℃反應2~10小時,反應結束后加飽和亞硫酸鈉溶液10.0~20.0毫升,加水10~30毫升稀釋,收集有機相并用無水硫酸鎂對其進行除水處理,過濾,真空濃縮,最后用10~30ml無水乙醇重結晶純化3次,過濾、干燥得化合物b:n,n,n′,n′-四(4-溴苯基)-1,4-苯二胺;
(ⅲ)在氬氣或氮氣保護下,在250ml三口瓶中分別加入n,n,n′,n′-四(4-溴苯基)-1,4-苯二胺、4-甲氧羰基苯硼酸、碳酸鉀和一定量的二氧六環,摩爾比n,n,n′,n′-四(4-溴苯基)-1,4-苯二胺:4-甲氧羰基苯硼酸甲酯:碳酸鉀為1:4.5~5.0:10.0~15.0,n,n,n′,n′-四(4-溴苯基)-1,4-苯二胺與溶劑的比為1毫摩:10.0~15.0毫升。加入一定量的鈀鹽作為催化劑,90℃反應20~30小時,反應結束蒸出1,4-二氧六環,加20~30ml水稀釋,二氯甲烷萃取(40~70ml×3),收集有機相并用無水亞硫酸鈉對其進行除水處理,過濾,蒸出二氯甲烷溶劑得粗產物,最后以石油醚和二氯甲烷按體積比為4:0.8~1.2的混合溶劑為洗脫劑用柱色譜法分離提純得到化合物c:n,n,n′,n′-四(4′-甲氧羰基聯苯基)-1,4-苯二胺;
(ⅳ)將n,n,n′,n′-四(4′-甲氧羰基聯苯基)-1,4-苯二胺和naoh按摩爾比1:30~50加入1,4-二氧六環和水的混合溶劑中,體積比為3~5:1,95℃回流8~12小時,蒸除去過量1,4-二氧六環,加水稀釋,加入過量的稀hno3進行酸化至ph=2,析出黃色固體,真空干燥得最終化合物d:n,n,n′,n′-四(4′-羧基聯苯基)-1,4-苯二胺。
所述的一種四元羧酸n,n,n′,n′-四(4′-羧基聯苯基)-1,4-苯二胺的合成方法,制備a時,在氮氣或氬氣保護下快速加入三-(2-甲苯基)膦,摩爾比1,4-二溴苯:二苯胺:叔丁醇鈉為1:2.1~2.3:2.1~2.5,1,4-二溴苯與甲苯的用量比是1毫摩:3.0~4.5毫升。
所述的一種四元羧酸n,n,n′,n′-四(4′-羧基聯苯基)-1,4-苯二胺的合成方法,制備a以甲苯作為溶劑,在85~100℃油浴加熱反應10~16小時,反應結束蒸出甲苯,加20~40ml水稀釋,二氯甲烷萃取(20~40ml×3),柱層析純化a以石油醚和乙酸乙酯按體積比為20:0.8~1.2。
所述的一種四元羧酸n,n,n′,n′-四(4′-羧基聯苯基)-1,4-苯二胺的合成方法,其特征在于:制備化合物a使用的催化劑為雙(二亞芐基丙酮)鈀,用量為1,4-二溴苯摩爾數的0.5~1.2%。
所述的一種四元羧酸n,n,n′,n′-四(4′-羧基聯苯基)-1,4-苯二胺的合成方法,制備化合物b時,a和nbs的摩爾比為1:3.5~5.5,需加50~80ml二氯甲烷作溶劑。
所述的一種四元羧酸n,n,n′,n′-四(4′-羧基聯苯基)-1,4-苯二胺的合成方法,其特征在于:制備化合物b時,純化b用10~30ml無水乙醇重結晶3次。
所述的一種四元羧酸n,n,n′,n′-四(4′-羧基聯苯基)-1,4-苯二胺的合成方法,制備化合物c時,摩爾比b:4-甲氧羰基苯硼酸:碳酸鉀為1:4.5~5.0:10.0~15.0。需加80~120ml二氧六環作溶劑。純化b用石油醚和二氯甲烷按體積比為4:0.8~1.2的混合溶劑為洗脫劑。
所述的一種四元羧酸n,n,n′,n′-四(4′-羧基聯苯基)-1,4-苯二胺的合成方法,制備化合物c使用的催化劑為四(三苯基膦)鈀,用量為b的物質的量的1~2.5%。
所述的一種四元羧酸n,n,n′,n′-四(4′-羧基聯苯基)-1,4-苯二胺的合成方法,n,n,n′,n′-四(4′-甲氧羰基聯苯基)-1,4-苯二胺和naoh按摩爾比1:30~50,溶劑1,4-二氧六環和水的混合溶劑中,體積比為3~5:1,酸化至ph=2。
用本方法合成結構新穎的n,n,n′,n′-四(4′-羧基聯苯基)-1,4-苯二胺的有益效果是:延長的剛性四羧酸類配體廣泛應用于mofs材料的合成中,這類材料通常具有有序的孔道結構、大的比表面積,使這類材料在氣體儲藏與分離、化學傳感、催化及藥物緩釋等不同領域具有潛在應用。延長的剛性四羧酸配體通常能得到結構新穎、比表面積大的多微孔mofs材料,并能改變微孔結構,使其氣孔內電子環境得到優化從而提高上述應用性能,因此,設計合成結構新穎的延長多羧酸配體并應用于mofs材料的合成已成為當今研究的熱點之一。然而,大尺寸的剛性多羧酸化合物合成比較困難。因此,用本方法合成結構新穎的n,n,n′,n′-四(4′-羧基聯苯基)-1,4-苯二胺是一種具有巨大潛在應用價值的四元羧酸配體。
附圖說明
圖1本發明中目標化合物d的結構式示意圖。
圖2本發明中化合物a和b的合成示意圖。
圖3本發明中化合物c和d的合成示意圖。
具體實施方式
本發明的n,n,n′,n′-四(4′-羧基聯苯基)-1,4-苯二胺的合成方法,合成步驟為:
以1,4-二溴苯,二苯胺為原料經buchwald-hartwig芳胺化反應生成n,n,n',n'-四苯基-1,4-苯二胺(a),a與nbs溴化反應生成n,n,n',n'-四(4-溴苯基)-1,4-苯二胺(b),合成路線如圖2所示。b與對甲氧羰基苯硼酸經suzuki偶聯反應生成n,n,n',n'-四(4'-甲氧羰基聯苯基)-1,4-苯二胺(c),c最后水解得到目標化合物n,n,n',n'-四(4'-羧基聯苯基)-1,4-苯二胺(d),合成路線如圖3所示。
具體實施例如下:
(ⅰ)如圖2所示:稱取1,4-二溴苯(2.9g,12.3mmol),二苯胺(5.0g,29.6mmol),雙(二亞芐基酮)鈀(0.04g,0.07mmol),叔丁醇鈉(2.9g,30.2mmol)于250ml三口瓶中,加入50ml甲苯,通入氬氣15min后迅速加入3.5ml三-(2-甲苯基)膦,氬氣保護下90℃油浴反應12h,反應結束蒸出甲苯,加30ml水稀釋,二氯甲烷萃取(30ml×3),收集有機層,無水na2so3干燥、過濾,蒸出溶劑得反應粗產物,干燥后經硅膠柱層析(洗脫劑:石油醚/乙酸乙酯=20:1)純化,得白色粉末a(3.04g,7.4mmol),產率60%。熔點:197~198℃。
化合物a核磁1hnmr、13cnmr和高分辨質譜數據:1hnmr(400mhz,cdcl3)δ7.28~7.22(m,8h),7.11(m,8h),6.98(m,8h).13cnmr(101mhz,cdcl3)δ142.82,132.12,129.15,125.42,123.70,122.38。hrms(esi),c30h24n2,實測值(計算值),m/z:413.5245[m+h]+(413.5250)。
(ⅱ)如圖2所示:稱取n,n,n',n'-四苯基-1,4-苯二胺(4.0g,9.7mmol)于250ml三口瓶中,緩慢向三口瓶中加入60ml二氯甲烷,加入nbs(6.9g,38.8mmol),室溫攪拌4h,停止反應,加入飽和na2so3溶液,分離有機相,無水mgso4干燥、過濾,蒸出氯仿得反應粗產物,無水乙醇重結晶3次,蒸餾水洗滌沉淀,得白色粉末b(6.9g,9.5mmol),產率,98%。熔點:99~101℃。
化合物b核磁1hnmr、13cnmr和高分辨質譜數據:1hnmr(400mhz,cdcl3)δ7.34(m,4h),7.26(m,4h),7.08(d,j=7.4hz,4h),6.95(m,8h).13cnmr(101mhz,cdcl3)δ:132.31,132.17,131.90,129.40,125.46,125.08。hrms(esi),c30h20br4n2,實測值(計算值),m/z:729.1084[m+h]+(729.1092)。
(ⅲ)如圖3所示:在250ml三口瓶中分別加入n,n,n',n'-四(4-溴苯基)-1,4-苯二胺(5.5g,7.6mmol)、4-甲氧羰基苯硼酸(6.6g,4.8eq),k2co3(13.8g,100.32mmol)和100ml1,4-二氧六環,通入n210min后,加2%四(三苯基膦)鈀,90℃反應24h,反應結束蒸出1,4-二氧六環,加30ml水稀釋,二氯甲烷萃取(60ml×3),收集有機層,無水na2so3干燥、過濾,蒸出二氯甲烷得反應粗產物,干燥后經硅膠柱層析(洗脫劑:石油醚/二氯甲烷=4:1)純化,得淡黃色粉末n,n,n',n'-四(4'-甲氧羰基聯苯基)-1,4-苯二胺(5.2g,5.5mmol),產率72%。熔點:345-347℃。
化合物c核磁1hnmr、13cnmr和高分辨質譜數據:1hnmr(400mhz,cdcl3)δ8.09(d,j=8.4hz,8h),7.99(d,j=8.2hz,8h),7.66(d,j=8.4hz,9h),7.51(d,j=2.1hz,4h),7.35(d,j=8.2hz,9h),3.96(s,12h).13cnmr(101mhz,cdcl3)δ166.97,144.59,142.97,139.63,133.07,131.58,130.15,129.33,129.06,127.69,126.31,117.85,52.27。hrms(esi),c62h48n2o8,實測值(計算值),m/z:959.0536[m+h]+(950.0531)。
(ⅳ)如圖3所示:在250ml的圓底燒瓶里依次加入n,n,n',n'-四(4'-甲氧羰基聯苯基)-1,4-苯二胺(3.5g,3.7mmol)、naoh(5g,125mmol)、h2o(20ml)和60ml1,4-二氧六環,95℃反應12h,蒸出1,4-二氧六環,加入適量的水溶解羧酸鈉鹽,過濾,濾液中加稀hno3酸化至ph為2,析出沉淀,靜置,傾倒上層清液,抽濾、干燥得目標化合物黃色沉淀d(3.23g,3.6mmol),產率98%。熔點:大于350℃。
化合物d核磁1hnmr、13cnmr和高分辨質譜數據:1hnmr(400mhz,dmso)δ12.96(s,4h),8.11-7.91(m,9h),7.85(m,2h),7.75(m,10h),7.56(m,9h),7.21-6.95(m,6h).13cnmr(101mhz,dmso)δ167.61,131.97,130.42,129.51,129.26,129.14,128.58,127.59,126.62,126.53,124.01。hrms(esi),c58h40n2o8[m-h]+,實測值(計算值),m/z:891.2805(891.2813)。
以上所述,僅是本發明的較佳實施例而已,并非對本發明作任何形式上的限制,任何未脫離本發明技術方案內容,依據本發明的技術實質對以上實施例所作的任何簡單修改、等同變化與修飾,均仍屬于本發明技術方案的范圍內。
- 還沒有人留言評論。精彩留言會獲得點贊!