一種高強高韌熱固性樹脂基復合材料的制備方法及應用與流程
本發明涉及一種高分子量嵌段共聚物ps-pcl-pdms-pcl-ps改性環氧樹脂的制備方法及應用。
背景技術:
環氧樹脂作為一類重要的熱固性樹脂,由于其優良的性能廣泛的應用于各個領域。但高交聯度帶來的固有脆性及差的阻尼性能限制了其在高性能復合材料中的應用。近年來,嵌段共聚物因其可以在選擇性溶劑中形成不同的形狀而廣受關注,在改性環氧樹脂方面也有研究,但很少有報道通過設計嵌段共聚物及其在環氧基體中的形態來獲得所需性能的研究。
殼核結構是一種比較經典的增韌環氧樹脂的方法。傳統的殼核結構是通過乳液聚合反應控制投料比來控制粒子的尺寸大小,一般選用橡膠態的柔性物質為核,剛性聚甲基丙烯酸甲酯(pmma)為殼,加入到環氧樹脂中可以有效的改善環氧樹脂的韌性,但其拉伸強度及模量會大大降低。
目前,衡正光等(hengz,zengz,zhangb,etal.enhancingmechanicalperformanceofepoxythermosetsviadesigningablockcopolymertoself-organizeinto“core–shell”nanostructure[j].rscadvances,2016,6(80):77030-77036.)人提供了一種低分子量的嵌段共聚物ps-pcl-pdms-pcl-ps(sldls-s)及環氧復合材料的制備方法。在增韌環氧樹脂的同時提升了復合材料的拉伸強度,但是效果并不明顯,有待進一步改進。
技術實現要素:
本發明通過控制反應時產物不同的投量比來制備高分子量的嵌段共聚物sldls(聚苯乙烯-聚己內酯-聚二甲基硅氧烷-聚己內酯-聚苯乙烯),將其加入到環氧樹脂中不僅提高了復合材料的拉伸強度,同時也提升了其韌性及動態剛性。
本發明提供了式ⅰ所示的嵌段共聚物,
所述嵌段共聚物n為18~645,t為10~706,m為5~100,數均分子量為50000~150000g/mol。
優選地,所述嵌段共聚物n為46~60,t為348~436,m為42,數均分子量為89162~104158g/mol。
優選地,所述嵌段共聚物的分子量分布指數為1~2,優選為1.23~1.52。
優選地,它是由如下原料制備而成:端羥基聚二甲基硅油、己內酯、2-溴異丁基酰溴、苯乙烯。進一步優選地,它是由如下原料制備而成:
1)由端羥基聚二甲基硅油和己內酯生成pcl-b-pdms-b-pcl,端羥基聚二甲基硅油和己內酯的摩爾比為1.36:(140~237);
2)pcl-b-pdms-b-pcl與2-溴異丁基酰溴反應,得br-pcl-b-pdms-b-pcl-br,pcl-b-pdms-b-pcl與2-溴異丁基酰溴的摩爾比為1:10;
3)br-pcl-b-pdms-b-pcl-br與苯乙烯反應,即得所述嵌段共聚物,br-pcl-b-pdms-b-pcl-br與苯乙烯的摩爾比為0.7:(540~567)。
本發明還提供了一種環氧樹脂復合材料,它是以前述的嵌段共聚物作為添加劑,加上環氧預聚體與固化劑,制備得到的環氧樹脂復合材料。
優選地,所述嵌段共聚物的添加量為環氧預聚體的1wt%~60wt%,優選為5wt%~20wt%。
優選地,環氧預聚體與固化劑的重量比為1.37:1~3:1。
本發明還提供了一種制備前述的嵌段共聚物的方法,其特征在于:它包括如下步驟:
(1)取端羥基聚二甲基硅油,與己內酯反應,得到pcl-b-pdms-b-pcl;
(2)將pcl-b-pdms-b-pcl與2-溴異丁基酰溴反應,得br-pcl-b-pdms-b-pcl-br;
(3)將br-pcl-b-pdms-b-pcl-br與苯乙烯反應,即得所述嵌段共聚物。
優選地,
步驟(1)的反應方法是:取端羥基聚二甲基硅油和干燥的甲苯在無水無氧瓶中共沸除水,蒸出多余的甲苯;然后將己內酯和辛酸亞錫加入到上述純化后的端羥基聚二甲基硅油中;經過三次液氮冷凍-真空脫氣-解凍循環后,將負壓狀態下的燒瓶置入120℃恒溫油浴中攪拌反應36h;待反應結束后,將粗產物溶解于二氯甲烷(dcm)溶液中,然后逐滴滴入冷凍甲醇中沉淀,過濾;優選地,所述辛酸亞錫的添加量為己內酯的1wt‰;
步驟(2)的反應方法是:將溶有三乙胺、pcl-b-pdms-b-pcl和4-二甲氨基吡啶的二氯甲烷溶液加入到三口圓底燒瓶中,在氬氣及冰浴的狀態下,攪拌混合均勻;將2-溴異丁基酰溴溶于二氯甲烷溶液中,冰浴狀態下通過恒壓漏斗逐滴加入到上述混合液中,滴加完成后室溫繼續攪拌反應24h;反應結束后將溶液旋蒸除去一半溶劑,將剩余產物逐滴滴入冷凍甲醇中沉淀,過濾;重復此溶解-沉淀-過濾過程三次,將純化后的產物在30℃的真空烘箱中干燥至恒重;
步驟(3)的反應方法是:向無水無氧瓶中依次加入br-pcl-b-pdms-b-pcl-br、苯乙烯單體、溴化亞銅、n,n,n’,n”,n”-五甲基二乙烯三胺,經過三次液氮冷凍-真空脫氣-解凍循環后,將負壓狀態下的燒瓶置入110℃恒溫油浴中磁力攪拌16h;待反應結束后,將產物暴露在空氣中冷卻至室溫至反應停止;將產物溶于dcm并通過中性氧化鋁柱子以除去催化劑;旋蒸濃縮溶液并逐滴滴入冷凍甲醇中沉淀,過濾;經三次溶解-沉淀-過濾過程后,將最終產物在30℃的真空烘箱中干燥至恒重,即可。
本發明制備得到了一種高分子量的嵌段共聚物,其與環氧樹脂的復合材料拉伸強度大,韌性好,動態剛性強,應用前景良好。
顯然,根據本發明的上述內容,按照本領域的普通技術知識和慣用手段,在不脫離本發明上述基本技術思想前提下,還可以做出其它多種形式的修改、替換或變更。
以下通過實施例形式的具體實施方式,對本發明的上述內容再作進一步的詳細說明。但不應將此理解為本發明上述主題的范圍僅限于以下的實例。凡基于本發明上述內容所實現的技術均屬于本發明的范圍。
附圖說明
圖1為根據本發明實施例1的嵌段共聚物ldl及sldls-l-1的nmr。
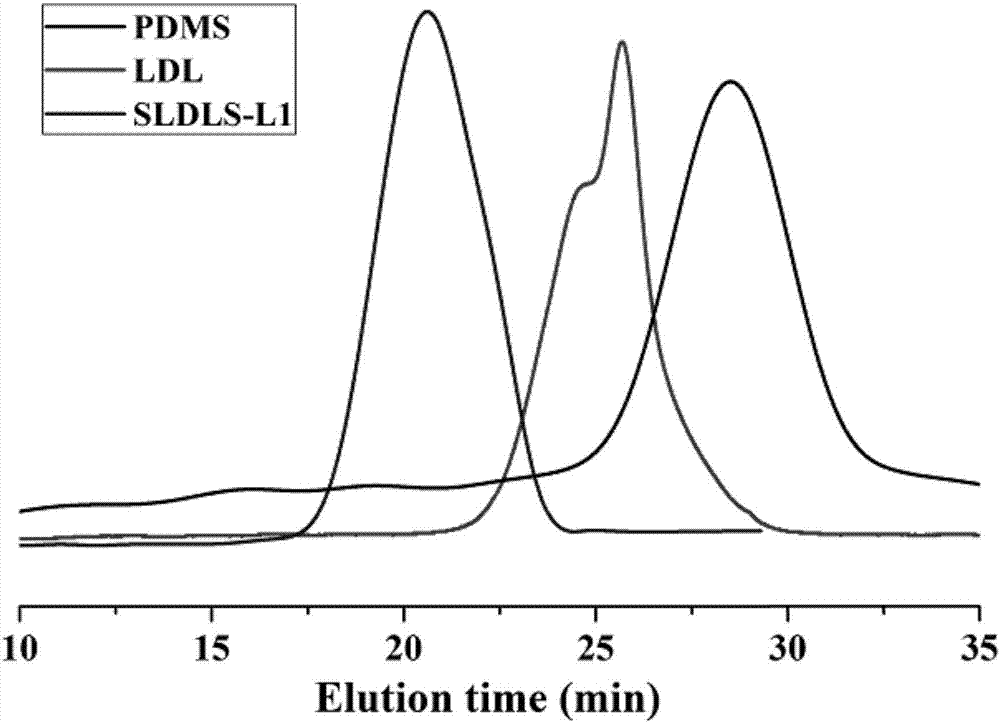
圖2為根據本發明實施例1的嵌段共聚物ldl及sldls-l-1的gpc。
圖3為根據本發明實施例2的嵌段共聚物ldl及sldls-l-2的nmr。
圖4為根據本發明實施例3的嵌段共聚物ldl及sldls-l-2的gpc。
圖5為根據本發明實施例3含10wt%(a,a’)和20wt%(b,b’)sldls-l-1的環氧樹脂的tem照片。
圖6不同材料的拉伸強度嵌段共聚物添加量的變化圖。
圖7不同材料的斷裂伸長率隨嵌段共聚物添加量的變化圖。
圖8不同材料的斷裂韌性(kic)隨嵌段共聚物添加量的變化圖。
具體實施方式
實施例1、高分子量嵌段共聚物ps-pcl-pdms-pcl-ps的制備(sldls-l1)
一、制備方法
高分子量嵌段共聚物sldls的合成過程如下:
1、pcl-b-pdms-b-pcl的制備
htpdms的分子量為3123,己內酯的分子量為114,2-溴異丁基酰溴的分子量為229.91,苯乙烯的分子量為104。
取4.234g端羥基聚二甲基硅油(htpdms,m=42)和干燥的甲苯在250ml無水無氧瓶中共沸除水,蒸出多余的甲苯,然后將16g己內酯(ε-cl)和辛酸亞錫[sn(oct)2](添加量為1wt‰ε-cl)加入到上述純化后的htpdms中。經過三次液氮冷凍-真空脫氣-解凍循環后,將負壓狀態下的燒瓶置入120℃恒溫油浴中攪拌反應36h。待反應結束后,將粗產物溶解于二氯甲烷(dcm)溶液中,然后逐滴滴入冷凍甲醇中沉淀,過濾。重復此溶解-沉淀-過濾過程三次以除去體系中可能存在的副產物和殘余單體。將純化后的產物在30℃的真空烘箱中干燥至恒重,產率為90~99%。得到的pcl-b-pdms-b-pcl的分子量為13582。
2、大分子引發劑br-pcl-b-pdms-b-pcl-br的制備
將溶有447mg三乙胺(tea)、15gho-pcl-b-pdms-b-pcl-oh和810mg4-二甲氨基吡啶(dmap)的dcm(二氯甲烷)溶液加入到三口圓底燒瓶中,在氬氣及冰浴的狀態下,攪拌混合均勻。將2.539g的2-溴異丁基酰溴(2-bib)溶于dcm溶液中,冰浴狀態下通過恒壓漏斗逐滴加入到上述混合液中,滴加完成后室溫繼續攪拌反應24h。反應結束后將溶液旋蒸除去一半溶劑,將剩余產物逐滴滴入冷凍甲醇中沉淀,過濾。重復此溶解-沉淀-過濾過程三次,將純化后的產物在30℃的真空烘箱中干燥至恒重,產率為95%~99%。
br-pcl-b-pdms-b-pcl-br的分子量為13732。
3、ps-b-pcl-b-pdms-b-pcl-b-ps的制備
向無水無氧瓶中依次加入10g大分子引發劑、66g苯乙烯單體、105mg溴化亞銅(cubr)、126mgn,n,n’,n”,n”-五甲基二乙烯三胺(pmdeta),經過三次液氮冷凍-真空脫氣-解凍循環后,將負壓狀態下的燒瓶置入110℃恒溫油浴中磁力攪拌16h。待反應結束后,將產物暴露在空氣中冷卻至室溫至反應停止。將產物溶于dcm并通過中性氧化鋁柱子以除去催化劑。旋蒸濃縮溶液并逐滴滴入冷凍甲醇中沉淀,過濾。經三次溶解-沉淀-過濾過程后,將最終產物在30℃的真空烘箱中干燥至恒重,產率為60~70%。
二、基本性質(ps-b-pcl-b-pdms-b-pcl-b-ps的化學結構及分子量檢測)
通過1hnmr及gpc對ps-b-pcl-b-pdms-b-pcl-b-ps的化學結構及分子量進行表征,結果如圖1及圖2所示。
結果表明:ps-b-pcl-b-pdms-b-pcl-b-ps成功合成,其數均分子量為104158,分子量分布指數1.52,其結構式為
其中,n為46,t為436,m為42。
實施例2、高分子量嵌段共聚物ps-pcl-pdms-pcl-ps的制備(sldls-l2)
一、制備方法
高分子量嵌段共聚物sldls的合成過程同實施例1。
htpdms的分子量為3123,己內酯的分子量為114,2-溴異丁基酰溴的分子量為229.91,苯乙烯的分子量為104。
1、pcl-b-pdms-b-pcl的制備
取4.234g端羥基聚二甲基硅油(htpdms,m=42)和一定量干燥的甲苯在250ml無水無氧瓶中共沸除水,蒸出多余的甲苯,然后將26.96g己內酯(ε-cl)和辛酸亞錫[sn(oct)2](添加量為1wt‰ε-cl)加入到上述純化后的htpdms中。經過三次液氮冷凍-真空脫氣-解凍循環后,將負壓狀態下的燒瓶置入120℃恒溫油浴中攪拌反應36h。待反應結束后,將粗產物溶解于適量的二氯甲烷(dcm)溶液中,然后逐滴滴入冷凍甲醇中沉淀,過濾。重復此溶解-沉淀-過濾過程三次以除去體系中可能存在的副產物和殘余單體。將純化后的產物在30℃的真空烘箱中干燥至恒重,產率為90~99%。得到的pcl-b-pdms-b-pcl的分子量為16874。
2、大分子引發劑br-pcl-b-pdms-b-pcl-br的制備
同實施例1。br-pcl-b-pdms-b-pcl-br的分子量為17024。
3、ps-b-pcl-b-pdms-b-pcl-b-ps的制備
向無水無氧瓶中依次加入12g、大分子引發劑56.896g苯乙烯單體、204mg溴化亞銅(cubr)、247mgn,n,n’,n”,n”-五甲基二乙烯三胺(pmdeta),經過三次液氮冷凍-真空脫氣-解凍循環后,將負壓狀態下的燒瓶置入110℃恒溫油浴中磁力攪拌16h。待反應結束后,將產物暴露在空氣中冷卻至室溫至反應停止。將產物溶于dcm并通過中性氧化鋁柱子以除去催化劑。旋蒸濃縮溶液并逐滴滴入冷凍甲醇中沉淀,過濾。經三次溶解-沉淀-過濾過程后,將最終產物在30℃的真空烘箱中干燥至恒重,產率為50~60%。
二、基本性質(ps-b-pcl-b-pdms-b-pcl-b-ps的化學結構及分子量檢測)
通過1hnmr及gpc對ps-b-pcl-b-pdms-b-pcl-b-ps的化學結構及分子量進行表征,結果如圖3及圖4所示。
結果表明:ps-b-pcl-b-pdms-b-pcl-b-ps成功合成,其數均分子量為89162,分子量分布指數1.23,其結構式為
其中,n為60,t為348,m為42。
實施例3、環氧樹脂基復合材料的制備
按表1所示比例,將實施例1制備的嵌段共聚物ps-b-pcl-b-pdms-b-pcl-b-ps(sldls-l1)加入到環氧預聚體(dgeba)中,在120℃下劇烈攪拌至形成均勻透明的溶液,再將固化劑moca加入體系中,快速劇烈攪拌直至獲得均勻透明的溶液。將所得共混溶液放入120℃真空烘箱1h以除去體系內氣泡,然后將其倒入聚四氟乙烯模具,150℃固化2h,然后升溫至180℃固化2h,固化反應結束后,脫模得到含嵌段共聚物的環氧熱固性樹脂。
實施例4環氧樹脂基復合材料的制備
按表1所示比例,將實施例2制備的嵌段共聚物ps-b-pcl-b-pdms-b-pcl-b-ps(sldls-l2)加入到環氧預聚體(dgeba)中,在120℃下劇烈攪拌至形成均勻透明的溶液,再將固化劑moca加入體系中,快速劇烈攪拌直至獲得均勻透明的溶液。將所得共混溶液放入120℃真空烘箱1h以除去體系內氣泡,然后將其倒入聚四氟乙烯模具,150℃固化2h,然后升溫至180℃固化2h,固化反應結束后,脫模得到含嵌段共聚物的環氧熱固性樹脂。
表1:不同混合液的用料配比
以下用試驗例的方式來說明本發明的有益效果:
試驗例1本發明制備的嵌段共聚物及環氧樹脂基復合材料所做的性質檢測
一、材料制備
1、本發明材料:分別取實施例3和實施例4制備的復合材料;
2、對比材料1:不含嵌段共聚物的環氧樹脂的制備
將60g環氧預聚體dgeba、24g固化劑moca在120℃下劇烈攪拌,直至形成均勻透明的溶液。將所得共混溶液放入120℃真空烘箱1h以除去體系內氣泡,然后將其倒入聚四氟乙烯模具,150℃固化2h,然后升溫至180℃固化2h,固化反應結束后,脫模得到不含嵌段共聚物的環氧熱固性樹脂
3、對比材料2:ldl/ep復合材料
制備方法為:按表2將嵌段共聚物pcl-b-pdms-b-pcl(ldl)加入到環氧預聚體(dgeba)中,在120℃下劇烈攪拌至形成均勻透明的溶液,再將固化劑moca加入體系中,快速劇烈攪拌直至獲得均勻透明的溶液。將所得共混溶液放入120℃真空烘箱1h以除去體系內氣泡,然后將其倒入聚四氟乙烯模具,150℃固化2h,然后升溫至180℃固化2h,固化反應結束后,脫模得到含嵌段共聚物的環氧熱固性樹脂。
表2:不同混合液的用料配比
4、對比材料3:含低分子量的嵌段共聚物的環氧樹脂的制備(sldls-s)
制備方法為:低分子量嵌段共聚物的制備方法同文獻hengz,zengz,zhangb,etal.enhancingmechanicalperformanceofepoxythermosetsviadesigningablockcopolymertoself-organizeinto“core–shell”nanostructure[j].rscadvances,2016,6(80):77030-77036,按表3將低分子量嵌段共聚物ps-b-pcl-b-pdms-b-pcl-b-ps(sldls-s)加入到環氧預聚體(dgeba)中,在120℃下劇烈攪拌至形成均勻透明的溶液,再將固化劑moca加入體系中,快速劇烈攪拌直至獲得均勻透明的溶液。將所得共混溶液放入120℃真空烘箱1h以除去體系內氣泡,然后將其倒入聚四氟乙烯模具,150℃固化2h,然后升溫至180℃固化2h,固化反應結束后,脫模得到含嵌段共聚物的環氧熱固性樹脂。
表3:不同混合液的用料配比
二、檢測方法
1、透射電鏡
利用徠卡uc7低溫超薄切片機在-120℃下對樣品進行切片,所得式樣厚度100~150nm。經過15min四氧化釕常溫熏染后,在tecnaig2f20型透射電鏡上進行觀察,低電子束流小于10ma,加速電壓為120kev。
由圖5可知,球狀嵌段共聚物均勻的分布在環氧樹脂基體內,尺寸為20~40nm。表明嵌段共聚物sldls-l1在環氧基體內發生微觀相分離,形成以柔性pdms為核、剛性ps為殼的“核-殼”結構,成功制備了含嵌段共聚物的環氧納米復合材料。
2、拉伸性能
采用instron5567萬能材料拉伸試驗機,根據gb/t2567-2008標準測試試樣的拉伸性能,測試速度為10mm/min。
由圖6可知,隨嵌段共聚物sldls含量的增加,納米復合材料的斷裂伸長率先增加后基本保持不變,拉伸強度先增加后降低。比如,當sldls-l1添加量為10wt%時,復合材料的拉伸強度達到最大值89.45mpa;當添加量為15wt%時,復合材料的斷裂伸長率達到最大值6.43%。
由圖7可知,相比于ldl改性環氧樹脂來說,sldls-l對復合材料的拉伸性能貢獻更大。當sldls-l嵌段共聚物的添加量為10wt%時,復合材料的拉伸強度和斷裂伸長率均高于ldl/ep復合材料。
3、動態機械性能分析
采用taq800動態機械性能測試儀的三點彎曲梯度升溫模式進行測試,升溫速率為3℃/min,測試溫度范圍為30~250℃。
表4為添加不同嵌段共聚物含量的復合材料的動態機械性能測試結果。隨著高分子量嵌段共聚物添加量的增加,復合材料的玻璃化轉變溫度逐漸降低,同時,復合材料的有效阻尼溫域明顯增加。加入高分子量嵌段共聚物,使復合材料的儲能模量增加,動態剛性增加。當添加量為15wt%時,復合材料的儲能模量達到最大值。
表4、不同嵌段共聚物含量的復合材料的動態機械性能測試
4、斷裂韌性分析
斷裂韌性測試樣條如圖所示。按照astme399標準,采用三點彎曲模式,測試速率為2mm/min。材料的斷裂韌性(kic)可由下式計算得到:
由圖8可知,嵌段共聚物的加入使環氧納米復合材料的kic值均高于純環氧樹脂且隨著嵌段共聚物含量的增加,kic值先增加后基本保持不變。比如,當sldls-l1嵌段共聚物添加量為20wt%時,復合材料的kic值達到1.43mn/m3/2。復合材料的斷裂韌性得到顯著提高。
三、總結
以雙端羥基聚二甲基硅氧烷,ε-己內酯和苯乙烯為原料,通過控制反應物不同的投料比,得到了一種既有柔性段(pcl和pdms)又有剛性段(ps)的兩親性高分子嵌段共聚物ps-b-pcl-b-pdms-b-pcl-b-ps。將上述嵌段共聚物與環氧樹脂共混,制備了具有納米結構的環氧樹脂復合材料。透射電鏡的結果表明嵌段共聚物在環氧樹脂基體內發生微相分離,嵌段共聚物自發形成的“殼-核”結構均勻分散在環氧樹脂基體內。
對納米復合材料進行的機械性能測試結果表明:相比于純環氧樹脂,嵌段共聚物的引入有效的提高了納米復合材料的拉伸強度、斷裂伸長率和斷裂韌性。動態機械性能分析結果表明,隨嵌段共聚物添加量的增加,復合材料的玻璃化轉變溫度逐漸降低,有效阻尼溫域和儲能模量明顯增加。斷裂韌性分析表明,嵌段共聚物的引入使納米復合材料的斷裂韌性得到顯著提高。
并且,與低分子量嵌段共聚物與環氧樹脂的復合材料相比,本發明高分子量嵌段共聚物與環氧樹脂的復合材料,其拉伸強度大幅提高,同時韌性和動態剛性也得到了提高。
綜上,本發明方法制備得到了一種高分子量的嵌段共聚物,還制備得到了它與環氧樹脂的復合材料,該復合材料的拉伸強度大,韌性好,動態剛性強,應用前景良好。
- 還沒有人留言評論。精彩留言會獲得點贊!