一類基于疊氮衍生物的可交聯太陽能電池界面材料的制作方法
本發明屬于功能高分子領域,具體涉及一類太陽能電池界面材料。
背景技術:
目前研究和應用最廣泛的太陽電池主要是單晶硅、多晶硅和非晶硅系列電池,然而該類電池具有原料成本高、生產工藝復雜、制造工藝成本高、能耗高、等缺點限制了其進一步產業化。作為第三代太陽能電池,有機太陽能電池具有低成本、柔性好、易制備等顯著優勢,通過調節材料結構來開發新型高轉化率的有機太陽能電池是一種可以有效提高太陽能電池的性能的研究方法。目前已有研究表明有機太陽能電池中,介于金屬氧化物和活性層之間的界面層,能夠有效改變金屬氧化物表面形貌、且對活性層縱向形貌有一定的調節作用。界面層對整個電池的能量轉化效率有著重要影響,對開發新型高轉化率的太陽能電池具有十分重要的意義。
近年來,科研人員設計合成了一系列基于9,9-二(烷基胺)芴片段的共聚物,能夠有效改進活性層與傳輸層的界面性能。曹鏞等人合成的pfn是目前廣泛使用的界面層材料,該材料可有效提高器件的voc,jsc,和ff[appl.phys.lett.2009,95,043301;j.mater.chem.,2010,20,2617–2622;adv.mater.2011,23,4636–4643;adv.funct.mater.2012,22,2846–2854;organicelectronics,2014,15,758–774]。針對該類材料與界面相互作用較弱的缺點,黃飛等人通過在脂溶性片段中引入烯烴[adv.energymater.2016,1502563]、氧丁環[j.am.chem.soc.,2013,135(41),15326–15329]等方式,在加熱或者光照條件下發生交聯來提高該類材料的穩定性及穩固與界面的相互作用。方俊峰等人報道了基于pfn氨基片段的小分子界面層材料[adv.energymater.2014,1400359],可將ptb7/pc71bm為活性層的太陽能電池效率提高到8.93%。彭強等人也報道了基于三苯胺結構的小分子受體材料[adv.funct.mater.2016,adfm.201504734],該類材料可將ptb7/pc71bm為活性層的太陽能電池效率提高到10.1%。
鑒于以上所述界面層對電池性能的影響,在界面層主鏈上引入疊氮衍生物制備的新型可交聯太陽能電池界面材料有望進一步改善金屬氧化物表面形貌,調節活性層縱向形貌分布,提升電荷傳輸能力,從而提高器件光伏性能,但是迄今還未見有關此類材料的制備方法及其應用于光伏電池中的報道。
技術實現要素:
為了克服現有技術存在的不足和缺陷,本發明提供一類基于疊氮衍生物的可交聯太陽能電池界面材料:具有疊氮可交聯的基于芴、茚并芴骨架的高分子聚合物,醇溶性好,熱穩定性好,可光照或加熱交聯。
將疊氮基團引入到聚合物烷基鏈末端,由于疊氮官能團在加熱或紫外光照下能夠交聯的特性,在極性片段修飾金屬氧化物表面、影響活性層縱向形貌分布的同時,通過聚合物鏈間交聯有效夠提升界面層穩定性,進一步提升器件光電轉化性能;該類材料可有效提升器件性能,表現出比目前廣泛使用的界面材料pfn更佳的光電轉化性能。
本發明的疊氮可交聯聚合物具有以下結構特征(式ⅰ~ⅵ):
式ⅰ~ⅵ中,x,y,z是各個單體之間摩爾數比例,滿足x=y+z,y=x+z,或者z=x+y特征之一;n為聚合度,是介于0~100萬之間的自然數;
界面親和單元pg選自下述結構之一:
該類結構包括氨基以其氮氧化物,與碳原子數為1到8的直鏈或支鏈的鹵代烷季銨化所得到的季銨鹽,與各類酸(包括但不限于硝酸根、乙酸根、硫酸根、磷酸根、高氯酸根等)所形成的鹽,或者氮氧化物等形態存在的各類衍生物。
脂溶性單元lg選自下述結構之一:
上述結構中:a1為c,si或者ge原子;a2為n,p,或者as原子;a3為o,s,或者se原子;alk為碳原子數為1-20的直鏈或支鏈的烷基;r1是氫原子,氟原子,碳原子數為1到20的直鏈或支鏈的烷基,或者末端為疊氮基的碳原子數為1-20的直鏈或支鏈的烷基;r2是氫原子,碳原子數為1到10的直鏈或支鏈的烷基;r3是氫原子,氟原子,碳原子數為1到20的直鏈或支鏈的烷基,或者末端為氨基及其烷基衍生物的碳原子數為1-20的直鏈或支鏈的烷基;r4為碳原子數為1-20的直鏈或支鏈的烷基;r5是氫原子,氟原子,碳原子數為1到20的直鏈或支鏈的烷基,碳原子數為1到20的直鏈或支鏈的烷氧基,碳原子數為1到20的直鏈或支鏈的鄰、間、對位烷基苯基,碳原子數為1到20的直鏈或支鏈的鄰、間、對位烷氧基苯基。
本發明提供優選的基于疊氮衍生物的可交聯太陽能電池界面材料,其結構式為如下化合物a-e中的任意一個:
結構式a
結構式b
結構式c
結構式d
結構式e
本發明的制備方法包括以下步驟:該反應在氮氣保護下進行,將xmol末端為溴、硼酸、或者硼酸酯中一種官能團的極性片段pg,ymol末端為溴、硼酸、或者硼酸酯中一種官能團的疊氮衍生物,zmol末端為溴、硼酸、或者硼酸酯中一種官能團的脂溶性片段lg按照x=y+z,y=x+z,或者z=x+y中一種比例加入到干燥的兩口燒瓶中,甲苯/四氫呋喃/氯仿/乙醚中的一種或幾種溶劑溶解,單體濃度控制在0.1mol/l左右,通惰性氣體0.5h后加入0.02倍摩爾量的pd催化劑,加入或者不加入堿性添加物氫氧化四乙胺/碳酸鉀/碳酸鈉/氫氧化鈉中的一種或多種。繼續通氣0.5h后開始加熱,回流反應24~48h后,停止反應。體系冷卻至室溫,將反應液滴入甲醇中沉降,過濾,收集的聚合物真空烘箱50℃烘12h,依次用甲醇、正己烷、氯仿進行索式提取,濃縮氯仿提取液,用甲醇再次沉降,過濾,得到如式i所示的含有茚并芴衍生物的共軛聚合物。
將該聚合物在四氫呋喃/甲醇/丙酮中的一種或幾種溶劑中,加入10當量的雙氧水氧化得到對應的氮氧化物。
將該聚合物在四氫呋喃/甲醇/丙酮中的一種或幾種溶劑中,加入10當量的r2x進行季銨化得到對應的季銨鹽類化合物。
將該聚合物在四氫呋喃/甲醇/丙酮中的一種或幾種溶劑中,加入10當量的質子酸(包括但不限于硝酸、乙酸、硫酸、磷酸、高氯酸等)得到對應的氨基鹽。
本發明能夠有效改善電極與活性層之間的電荷傳輸性能,降低電極表面功函數;通過紫外光照或加熱后交聯可進一步提高界面層穩定性;可作為界面修飾材料在太陽能電池等領域中應用。
本發明的主要優點在于:
1.合成的基于疊氮衍生物的可交聯太陽能電池界面材料可通過正交溶液加工,易溶于甲醇,乙醇,水等極性溶劑;
2.合成的基于疊氮衍生物的可交聯太陽能電池界面材料熱穩定性好,其實分解溫度大于300℃;
3.合成的基于疊氮衍生物的可交聯太陽能電池界面材料,能夠有效改善電極與活性層之間的電荷傳輸性能,降低電極表面功函數,適合做界面材料;
4.合成的基于疊氮衍生物的可交聯太陽能電池界面材料,可通過紫外光照或加熱后交聯可進一步提高界面層穩定性;
5.合成的基于疊氮衍生物的可交聯太陽能電池界面材料作為界面層在太陽能電池中展示出了非常顯著的光電轉化效率提升,優于目前廣泛使用的界面材料pfn。
6.制備的基于疊氮衍生物的可交聯太陽能電池界面材料,成本低廉、性能優異、穩定性高,適合大面積制備使用。
附圖說明
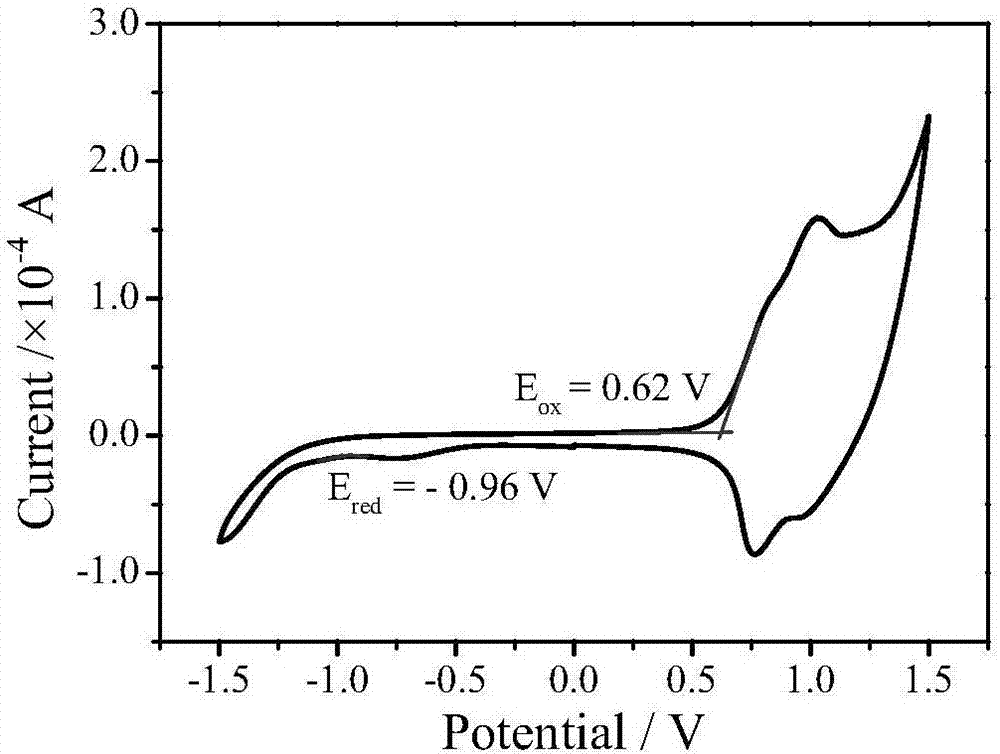
圖1是a~e中某一個共軛聚合物的c-v圖。
圖2是a~e中某一個共軛聚合物的c-v圖。
圖3是a~e中某一個共軛聚合物的c-v圖。
圖4是本發明的聚合物界面層光伏電池的結構示意圖。
圖5是zno、pfn或結構式為a~e中某一個共軛聚合物為界面層時,使用富勒烯受體材料pc71bm時光伏電池的j-v曲線。
具體實施方式
下面分別對聚合物、聚合前體的合成方法進行詳細闡述,所舉例子并不限制本專利保護范圍,只是為了更好地理解本發明。用元素分析、核磁共振、質譜等表征本發明疊氮可交聯聚合物的化學結構,用熱衷分析表征其熱穩定性,用循環伏安法表征其電化學性質,用紫外吸收光譜研究其光物理性質。
實施例1:脂溶性單元的合成
(1)6,6,12,12-四正辛基茚[1,2-b]芴(5)的制備
2.54g化合物4(10mmol)于100ml四氫呋喃/乙醚/甲基叔丁基醚中某一種或幾種溶劑中,ar保護,-20℃下滴加n-buli(2.5m,30mmol,12ml)并緩慢升至室溫后攪拌1.5h,再次冷卻至-20℃下加入正辛基溴(30mmol)后升至室溫下攪拌4h。再次加入30mmoln-buli和30mmol正辛基溴后于室溫下反應過夜并點板檢測反應。萃取,旋干溶劑后加入石油醚析出固體,柱層析收白色固體8.7g,收率95%。1hnmr(500mhz,cdcl3),δ(ppm):7.78(dd,j=2.0,7.0hz,2h),7.56(s,2h),7.37(dd,j=3.0,13.5hz,4h),7.34-7.27(m,2h),2.08-2.04(m,8h),1.21-1.08(m,40h),0.83(t,j=7.0hz,12h),0.71-0.69(m,8h).13cnmr(125mhz,cdcl3),δ(ppm):151.1,150.0,141.5,140.5,126.6,122.8,119.3,113.8,54.7,40.7,31.8,30.0,29.2,29.2,23.7,22.6,14.0.ms:702.6.
(2)2,8-二溴-6,6,12,12-四正辛基茚[1,2-b]芴(6)的制備
1.9g化合物5(27mmol)于30ml苯/四氯化碳/dmf/dmso中的一種或幾種溶劑中,加入br2/cubr/cubr2中的一種或幾種作為溴源,加熱到特定溫度下反應24h。過濾,旋干濾液得到灰白色固體,過柱純化得2.1g白色固體,收率90%。1hnmr(500mhz,cdcl3),δ(ppm):7.59(dd,j=2.0,7.0hz,2h),7.55(s,2h),7.47-7.45(m,4h),1.98(t,j=7.5hz,8h),1.18-1.03(m,40h),0.79(t,j=7.0hz,12h),0.63-0.61(m,8h).13cnmr(125mhz,cdcl3),δ(ppm):153.4,150.0,140.3,140.0,129.8,126.1,120.9,120.8,114.0,55.1,40.5,31.8,29.9,29.2,29.2,23.7,22.6,14.1.ms:1011.2.
(3)偶聯前體化合物7的制備
10mmol聚合前體6在惰性氣體保護下,溶于50ml四氫呋喃/乙醚/甲基叔丁基醚中某一種溶劑或者某幾種溶劑的混合中,在低溫下加入4當量正丁基鋰后于低溫下反應1h,再加入3當量的2-異丙氧基-4,4,5,5-四甲基-1,3,2-二雜氧戊硼烷或者三甲基氯化錫后升至室溫下反應24h。過柱純化得到白色固體為所需聚合前體。以化合物6‘為例,其表征數據具有如下特征:1hnmr(500mhz,cdcl3),δ(ppm):7.81(d,j=7.5hz,2h),7.74(dd,j=4.5,7.0hz,4h),7.63(s,2h),2.06-1.99(m,8h),1.40(s,24h),1.16-1.05(m,40h),0.77(t,j=7.0hz,12h),0.62-0.60(m,8h).13cnmr(125mhz,cdcl3),δ(ppm):150.6,150.4,144.5,140.9,133.7,128.8,118.8,114.4,83.7,54.8,40.5,31.8,30.0,29.2,29.2,25.0,23.7,22.6,14.1.ms:955.1.
實施例2:界面親和單元的合成
(1)2,7-二溴-9,9-二(6-二甲氨基己基)芴(12)的制備
1,6-二溴己烷(40ml,256mmol),40ml的koh(50%)溶液和四丁基溴化銨(tbab,1.436g,4.3mmol)依次加入反應瓶中,升溫至75℃下加入化合物11(5g,15.4mmol)繼續反應15min,冷至室溫下用氯仿萃取(30mlx3),有機相用1mhcl洗,飽和食鹽水洗,無水硫酸鎂干燥,除去dcm后減壓蒸餾除去1,6-二溴己烷,粗產物用硅膠柱純化(pe)。
將上述產物溶于50ml四氫呋喃/乙醚/甲基叔丁基醚中某一種溶劑或者某幾種溶劑的混合中,加入20當量的二甲胺水溶液避光反應24h后得到所需化合物,過柱純化得到白色固體。1hnmr(500mhz,cdcl3),δ(ppm):7.55(d,j=9.0hz,2h),7.47-7.45(m,4h),2.81(dd,j=8.5,11.0hz,4h),2.68(s,12h),1.94-1.91(m,4h),1.60-1.57(m,4h),1.16-1.11(m,4h),0.61-0.58(m,4h).13cnmr(125mhz,cdcl3),δ(ppm):152.1,139.0,130.4,126.1,121.5,121.4,58.2,55.5,43.4,39.7,34.8,29.0,26.1,24.6,23.2.ms:578.5.
(2)2,7-二(4,4,5,5-四甲基-1,3,2-二雜氧戊硼烷基)-9,9-二(6-二甲氨基己基)芴(13)的制備
10mmol化合物12在惰性氣體保護下,溶于50ml四氫呋喃/乙醚/甲基叔丁基醚中某一種溶劑或者某幾種溶劑的混合中,在低溫下加入4當量正丁基鋰后于低溫下反應1h,再加入3當量的2-異丙氧基-4,4,5,5-四甲基-1,3,2-二雜氧戊硼烷后升至室溫下反應24h。過柱純化得到白色固體為所需聚合前體。1hnmr(500mhz,cdcl3),δ(ppm):7.82(d,j=7.5hz,2h),7.76(d,j=7.5hz,2h),2.84(dd,j=8.5,11.0hz,4h),2.68(s,12h),1.94-1.91(m,4h),1.60-1.57(m,4h),1.42(s,24h),1.16-1.11(m,4h),0.61-0.58(m,4h).13cnmr(125mhz,cdcl3),δ(ppm):150.1,138.0,130.7,126.9,121.6,121.5,83.7,58.8,55.1,43.2,39.5,34.1,29.4,26.6,25.2,24.2,23.4.ms:672.6.
實施例3:含有疊氮官能團的偶聯前體合成
x為溴、硼酸、硼酸酯中一種。
15.4mmol化合物1于150ml新開瓶的dmso中,n2保護下加入疊氮化鈉(3eq.,46.2mmol,3g)并升溫至70℃下反應12h,tlc檢測完全反應后倒入500ml冰水中,氯仿萃取三次,合并有機相,無水硫酸鎂干燥。旋干得到所需化合物。hplc>99%。以x=br為例:1hnmr(500mhz,cdcl3),δ(ppm):7.52(d,j=8.0hz,2h),7.47-7.43(m,2h),7.26(s,2h),3.15(t,j=7.0hz,4h),1.94-1.91(m,4h),1.44-1.38(m,4h),1.17-1.05(m,8h),0.62-0.60(m,4h).
實施例4:以結構式為a的共軛聚合物的合成為例
x1、x2、x3為溴、硼酸、硼酸酯中一種,三者不同時為溴或者硼的衍生物。
在氬氣保護下,將1.0mmol雙臂茚并芴衍生物1、0.8mmol界面親和單元2和0.2mmol疊氮衍生物3加入到干燥的兩口燒瓶中,用10ml甲苯/四氫呋喃/氯仿/乙醚中的一種或幾種溶解,通氬氣0.5h后加入0.02當量的pd催化劑,加入或者不加入堿性添加物氫氧化四乙胺/碳酸鉀/碳酸鈉/氫氧化鈉中的一種或多種,繼續通氣0.5h,然后加熱回流24~48h。停止反應。體系冷卻至室溫,加入20ml水淬滅反應,用氯仿(20ml×3)萃取,合并有機相并于無水硫酸鈉干燥。旋干溶劑后加入少量氯仿溶解,將其滴入甲醇中沉降,過濾,收集的聚合物真空烘箱50℃烘12h,依次用甲醇、正己烷、氯仿進行索式提取,濃縮氯仿提取液,用甲醇再次沉降,過濾,得到聚合物a。
實施例5:以結構式為b的共軛聚合物的合成為例
x1、x2、x3為溴、硼酸、硼酸酯中一種,三者不同時為溴或者硼的衍生物。
在氬氣保護下,將1.0mmol雙臂茚并芴衍生物1、0.9mmol界面親和單元2和0.1mmol疊氮衍生物3加入到干燥的兩口燒瓶中,用10ml甲苯/四氫呋喃/氯仿/乙醚中的一種或幾種溶解,通氬氣0.5h后加入0.02當量的pd催化劑,加入或者不加入堿性添加物氫氧化四乙胺/碳酸鉀/碳酸鈉/氫氧化鈉中的一種或多種,繼續通氣0.5h,然后加熱回流24~48h。停止反應。體系冷卻至室溫,加入20ml水淬滅反應,用氯仿(20ml×3)萃取,合并有機相并于無水硫酸鈉干燥。旋干溶劑后加入少量氯仿溶解,將其滴入甲醇中沉降,過濾,收集的聚合物真空烘箱50℃烘12h,依次用甲醇、正己烷、氯仿進行索式提取,濃縮氯仿提取液,用甲醇再次沉降,過濾,得到聚合物b。
實施例6:以結構式為c的共軛聚合物的合成為例
x1、x2、x3為溴、硼酸、硼酸酯中一種,三者不同時為溴或者硼的衍生物。
在氬氣保護下,將1.0mmol雙臂茚并芴衍生物1、0.95mmol界面親和單元2和0.05mmol疊氮衍生物3加入到干燥的兩口燒瓶中,用10ml甲苯/四氫呋喃/氯仿/乙醚中的一種或幾種溶解,通氬氣0.5h后加入0.02當量的pd催化劑,加入或者不加入堿性添加物氫氧化四乙胺/碳酸鉀/碳酸鈉/氫氧化鈉中的一種或多種,繼續通氣0.5h,然后加熱回流24~48h。停止反應。體系冷卻至室溫,加入20ml水淬滅反應,用氯仿(20ml×3)萃取,合并有機相并于無水硫酸鈉干燥。旋干溶劑后加入少量氯仿溶解,將其滴入甲醇中沉降,過濾,收集的聚合物真空烘箱50℃烘12h,依次用甲醇、正己烷、氯仿進行索式提取,濃縮氯仿提取液,用甲醇再次沉降,過濾,得到聚合物c。
實施例7:以結構式為d的共軛聚合物的合成為例
聚合物a(100mg)溶于在四氫呋喃/甲醇/丙酮中的一種或幾種溶劑中,加入0.8ml溴乙烷后避光反應24h,旋干溶劑即得到所需的聚合物d。
實施例8:以結構式為e的共軛聚合物的合成為例
x1、x2、x3為溴、硼酸、硼酸酯中一種,三者不同時為溴或者硼的衍生物。
在氬氣保護下,將0.9mmol雙臂茚并芴衍生物1、1.0mmol界面親和單元2和0.1mmol疊氮衍生物3加入到干燥的兩口燒瓶中,用10ml甲苯/四氫呋喃/氯仿/乙醚中的一種或幾種溶解,通氬氣0.5h后加入0.02當量的pd催化劑,加入或者不加入堿性添加物氫氧化四乙胺/碳酸鉀/碳酸鈉/氫氧化鈉中的一種或多種,繼續通氣0.5h,然后加熱回流24~48h。停止反應。體系冷卻至室溫,加入20ml水淬滅反應,用氯仿(20ml×3)萃取,合并有機相并于無水硫酸鈉干燥。旋干溶劑后加入少量氯仿溶解,將其滴入甲醇中沉降,過濾,收集的聚合物真空烘箱50℃烘12h,依次用甲醇、正己烷、氯仿進行索式提取,濃縮氯仿提取液,用甲醇再次沉降,過濾,得到聚合物e。
實施例9:上述界面材料的電化學測試
用chi660d型電化學工作站,采用玻碳為工作電極,鉑絲電極為對電極,ag/ag+電極為參比電極,bu4n·pf6作電解質,在乙腈/甲醇=1:5的溶劑中,經循環伏安法測定各聚合物的homo和lumo能級。
實施例10:上述界面材料在光伏器件中的應用
采用如附圖3所示的三明治型電池結構,將聚合物a-e按一定濃度溶于甲醇/乙酸/甲苯/四氫呋喃的一種或者幾種溶液中,按照一定厚度旋涂于zno表面;活性層采用ptb7-th與小分子受體按照一定的重量比制成活性層,有效面積0.16cm2,在newportthermaloriel69911模擬太陽光源下進行電流-電壓測試,利用keithley2400源表采集。部分界面材料的器件結果見表1,以pfn為界面層的i-v曲線和eqe見附圖4,以a~p為界面層的部分i-v曲線和eqe見附圖5。與pfn相比,聚合物a~p為界面層時可在不改變開路電壓(voc=0.98v)的前提下有效提高短路電流(jsc=12.37ma/cm2,相比于pfn為界面層的器件提高0.26ma/cm2),填充因子ff也有所提高。綜合器件性能可有效提高0.2%~1.0%。
表1是不同器件結構、不同膜厚下,zno、pfn或結構式為a~e中某一個共軛聚合物為界面層,使用富勒烯受體材料pc71bm時光伏電池性能數據。
表1
溶劑:氯苯+對二碘苯(97.5:2.5);
器件結構:ito/zno/activelayer(90±0nm)/moo3(10nm)/al(100nm);
活性層:ptb7:pc71bm=1:1.510mg/ml;
器件面積0.16cm2。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 第9族過渡金屬催化劑及其使用方法
- 一種(s)-(+)-1-(2-哌啶苯基)-3-甲基正丁胺的制備方法
- 感放射線性樹脂組合物、硬化膜及其形成方法以及顯示元件的制作方法
- 一種3-疊氮-2-芳基丙腈的合成方法
- 一種以疊氮化丙烯酰氯-苯乙烯共聚物改性碳納米管為抗靜電劑的聚苯乙烯超微粉體的 ...的制作方法
- 用于涂料、粘合劑、密封劑和彈性體應用的聚氨酯聚合物經銅疊氮化物-炔烴點擊化學的合成的制作方法
- 具有改進的可加工性的基于乙烯的聚合物組合物的制作方法
- Rgd和/或ogp10-14的1,4,5-三取代的1,2,3-三唑模擬物、其獲得方法及其用圖
- 一種三組分一鍋法合成1,2,3-三氮唑類化合物的方法
- 一種汽車安全氣囊用煙火式氣體發生器的制造方法