有效光降解偶氮染料的新型稀土鐠配合物及其制備方法和應用與流程
本發明涉及鐠金屬配合物,具體地,涉及有效光降解偶氮染料的新型稀土鐠配合物及其制備方法和應用。
背景技術:
配位聚合物又稱為金屬—有機骨架化合物(metal-organicframeworks,縮寫成mofs),是指由金屬原子或者原子簇與有機配體通過共價鍵、配位鍵和分子間的相互作用等聚合而成的具有周期性網狀結構的晶體材料。
稀土配位化學是稀土化學領域的研究熱點之一,稀土就是元素周期表中的鑭系元素,其性質較軟,在空氣中易潮解,大多數元素價態為+3價,可形成穩定的配合物。稀土配合物是稀土金屬離子與含氧,氮有機配體通過配位鍵合作用形成的配合物,稀土配合物難生成,但稀土配合物具有豐富而新穎的結構,同時其獨特的功能性也表明著配合物在光、磁、光催化等諸多領域具有潛在的研究價。稀土鐠是稀土元素中用量較大的元素之一,在空氣中抗腐蝕能力較其他稀土金屬元素強,含氮、氧元素的稀土鐠配合物具有良好的光學性能,在光學材料、檢測重金屬離子、光催化等領域有很好的應用前景。
目前,有機染料的降解一般是使用半導體材料、無機復合等材料光催化降解染料,但是稀土有機配合物材料用于光催化降解染料的報道很少,同時利用半導體及其半導體復合材料降解染料存在催化劑難分離,不易固定等不足之處。
技術實現要素:
本發明的目的是提供一種有效光降解偶氮染料的新型稀土鐠配合物及其合成方法,該新型稀土鐠配合物具有良好的穩定性以及光催化降解偶氮染料(mb)性能,同時該制備方法簡單,而且解決了染料難降解的問題。
為了實現上述目的,本發明提供了一種有效光降解偶氮染料的新型稀土鐠配合物,該新型稀土鐠配合物分子式為:
{[pr3+(l2-)1.5(dmf)(h2o)2](dmf)}n,其中,l2-為2,2'-聯吡啶-4,4'-二羧酸脫去兩個質子后的基團,dmf為n,n-二甲基甲酰胺,n為正整數。
本發明還提供了一種上述制備新型稀土鐠配合物的方法,該制備方法為:以n,n-二甲基甲酰胺與水的混合溶液為溶劑體系中,將鐠鹽、h2l、4,4'-雙(1h-咪唑-1-取代)-1,1'-聯苯進行配位反應以制得稀土鐠配合物;其中,h2l為2,2'-聯吡啶-4,4'-二羧酸。
本發明進一步地提供了一種有效光降解偶氮染料的新型稀土鐠配合物在光催化降解mb染料中的應用。
在上述技術方案中,本發明通過溶劑熱法制備了一種有效光降解偶氮染料的新型稀土鐠配合物,具體為:以n,n-二甲基甲酰胺與水的混合溶液為溶劑體系、鐠鹽為金屬源、h2l為主配體、4,4'-雙(1h-咪唑-1-取代)-1,1'-聯苯為輔配體(輔配體的作用是改變配合物空間的延伸方向,使結構多樣化)進行配位反應,該制備方法將對稀土離子具有配位作用的吡啶羧酸型配基引入聚合物側基,然后與與pr3+配位,得到效光降解偶氮染料的新型稀土鐠配合物。該新型稀土鐠配合物的光催化降解性能優異且結構穩定,進而使其能夠應用于光催化降解染料。同時該制備方法簡單、產率高、而且解決了染料難降解的問題。
本發明的其他特征和優點將在隨后的具體實施方式部分予以詳細說明。
附圖說明
附圖是用來提供對本發明的進一步理解,并且構成說明書的一部分,與下面的具體實施方式一起用于解釋本發明,但并不構成對本發明的限制。在附圖中:
圖1是檢測例1中晶體結構不對稱單元的橢球圖;
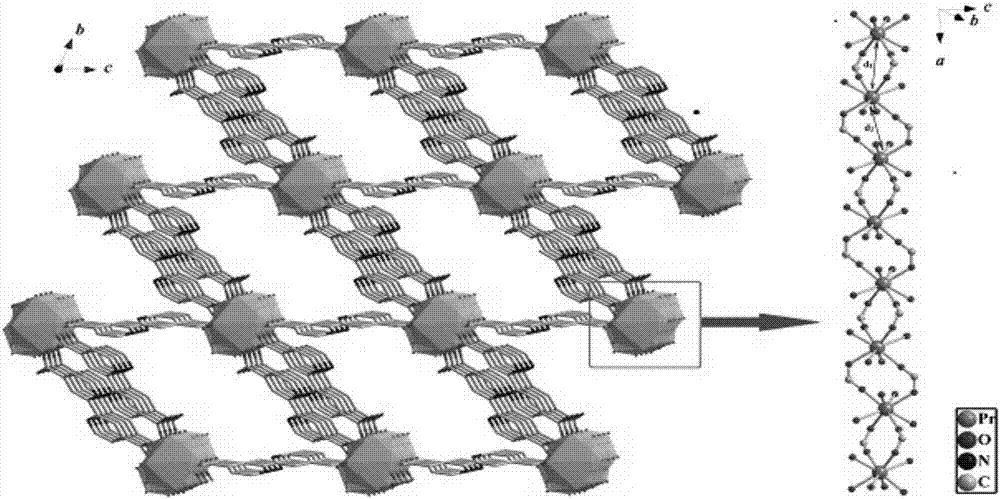
圖2為檢測例1中新型鐠配合物的空間三維圖;
圖3為檢測例3中新型鐠配合物的熱重分析圖;
圖4為檢測例4中新型鐠配合物的固體紫外可見光譜圖;
圖5為檢測例4中新型鐠配合物的光能對應于能量的kubelka-munk函數的變形圖;
圖6為檢測例5中新型鐠配合物的紅外發射光譜圖;
圖7為檢測例6中新型鐠配合物的x-射線粉末衍射譜圖;
圖8是實施例1的新型鐠配合物的在不同條件下對偶氮染料的催化活性譜圖;
圖9為實施例1的新型鐠配合物作為催化劑,紫外光照下的紫外-可見吸收光譜圖;
圖10為實施例1的新型鐠配合物作為催化劑,可見光照下的紫外-可見吸收光譜圖;
圖11為實施例1的新型鐠配合物作為催化劑,無光照下的紫外-可見吸光譜圖;
圖12為無催化劑,紫外光照下的紫外-可見吸收光譜圖。
具體實施方式
以下對本發明的具體實施方式進行詳細說明。應當理解的是,此處所描述的具體實施方式僅用于說明和解釋本發明,并不用于限制本發明。
在本文中所披露的范圍的端點和任何值都不限于該精確的范圍或值,這些范圍或值應當理解為包含接近這些范圍或值的值。對于數值范圍來說,各個范圍的端點值之間、各個范圍的端點值和單獨的點值之間,以及單獨的點值之間可以彼此組合而得到一個或多個新的數值范圍,這些數值范圍應被視為在本文中具體公開。
本發明提供了一種有效光降解偶氮染料的新型稀土鐠配合物,該新型稀土鐠配合物分子式為:{[pr3+(l2-)1.5(dmf)(h2o)2](dmf)}n,其中,l2-為2,2'-聯吡啶-4,4'-二羧酸脫去兩個質子后的基團,dmf為n,n-二甲基甲酰胺,n為正整數。
在上述有效光降解偶氮染料的新型稀土鐠配合物中,各元素之間的連接關系以及位置關系可以在寬的范圍內選擇,但是為了使得新型稀土鐠配合物具有更優異的光催化以及穩定性,優選地,新型稀土鐠配合物晶體的配位方式滿足以下條件:pr3+離子存在于配位環境,pr3+為稍微扭曲的八面體配位構型;每個pr(iii)離子都與八個氧原子配位,其中五個氧原子來自l2-配體,另一個氧原子來自dmf分子,最后兩個氧原子來自水分子。
在上述配合物中,金屬離子m(pr3+)與主配體h2l的配位方式可以是多種,可以是式(a)-(e)中的任意一種,
在上述新型稀土鐠配合物為晶體的情形下,該新型稀土鐠配合物的晶型條件可以在寬的范圍內選擇,但是為了使得新型稀土鐠配合物具有更優異的光催化以及穩定性,優選地,稀土鐠配合物的晶型滿足以下條件:三斜晶系,p-1空間群,晶胞參數分別為:
本發明還提供了一種上述的新型稀土鐠配合物的制備方法,該制備方法為:以n,n-二甲基甲酰胺與水的混合溶液為溶劑體系中,將鐠鹽、h2l、4,4'-雙(1h-咪唑-1-取代)-1,1'-聯苯進行配位反應以制得新型稀土鐠配合物;其中,h2l為2,2'-聯吡啶-4,4'-二羧酸。
在上述制備方法中,配位反應的具體條件可以在寬的范圍內選擇,但是為了使得制得的新型稀土鐠配合物具有更優異的光催化以及穩定性,同時使得制備方法具有更優異的產率,優選地,配位反應至少滿足以下條件:反應溫度為60-75℃,反應時間為35-50h。
在上述制備方法中,各物料的用量可以在寬的范圍內選擇,但是為了使得制得的新型稀土鐠配合物具有更優異的光催化以及穩定性,同時使得制備方法具有更優異的產率,優選地,相對于0.1mmol的所述鐠鹽,所述溶劑體系的用量為4-6ml;其中,鐠鹽、h2l、4,4'-雙(1h-咪唑-1-取代)-1,1'-聯苯的摩爾比為12:1-2.5:0.8-2,n,n-二甲基甲酰胺與水的體積比為0.6:2.8-4.5;更優選地,鐠鹽、h2l、4,4'-雙(1h-咪唑-1-取代)-1,1'-聯苯的摩爾比為12:1.4-1.8:0.9-1.1,n,n-二甲基甲酰胺與水的體積比為0.6:3.8-4.2。
在上述制備方法中,鐠鹽的具體種類可以在寬的范圍內選擇,但是從鐠鹽溶解性以及成本上考慮,優選地,鐠鹽為pr(no3)3·6h2o、pr2(so4)3·8h2o和prcl3·6h2o的至少一者。
在上述實施方式的基礎上,為了使新型稀土鐠配合物具有更高的產率,優選地,在配位反應之前,該制備方法還包括添料工序,具體為:首先添加所述鐠鹽、h2l、4,4'-雙(1h-咪唑-1-取代)-1,1'-聯苯,然后添加所述溶劑體系,最后在25-30℃下超聲震蕩3-10min使主輔配體和金屬鹽完全溶解;最后將其上述混合溶液裝入以聚四氟乙烯作襯里的不銹鋼反應釜中反應;由此通過在配位反應之前將各物料之間充分的混合進而能夠起到提高產率的作用。
同時,為了進一步提高新型稀土鐠配合物的產率,優選地,在配位反應之前,該制備方法還包括后處理工序,具體為:將反應體系自然冷卻至31-36℃,接著進行固液分離,然后將母液多次洗滌固液分離所得到的固體,再于25-30℃下自然干燥得到少量深綠色立方晶體,即新型稀土鐠配合物。通過該后處理能夠使得配合物能夠形成晶體自反應體系中析出,進而提高稀土鐠配合物的產率。
本發明進一步地提供了一種如上述的新型稀土鐠配合物在光催化降解偶氮染料染料中的應用。
在上述應用中,偶氮染料的具體結構可以在寬的范圍內選擇,但是從偶氮染料的使用程度以及市場占有率考慮,優選地,所述偶氮染料可以是如甲基橙染料,還可以是剛果紅染料。
在上述應用中,具體的操作方式可以有多種,但是為了進一步提高催化降解效率,優選地,光催化降解的具體步驟為:先將新型稀土鐠配合物與偶氮染料染料溶液于陰暗的條件下攪拌0.5-1h(使得催化劑與染料之間達到吸附-解析平衡),接著在紫外線存在的條件下進行進行降解反應40-80min。
此外,在上述應用中,新型稀土鐠配合物的用量可以在寬的范圍內選擇,但是為了進一步提高降解效率以及成本上考慮,優選地,相對于30ml的染料溶液,稀土鐠配合物光催化材料的用量為15-20mg。
以下將通過實施例對本發明進行詳細描述。
實施例1
1)將0.12mmolpr(no3)3·6h2o、0.0168mmol2,2'-聯吡啶-4,4'-二羧酸、0.008mmol4,4'-雙(1h-咪唑-1-取代)-1,1'-聯苯的混合物,裝入以聚四氟乙烯內膽中,然后加入5ml的n,n-二甲基甲酰胺與水的混合溶液(n,n-二甲基甲酰胺與水的體積比為1:4),25℃下超聲震蕩5min;
2)將裝有上述混合溶液的反應釜放入烘箱中,在60℃下恒溫反應48h后自然降溫至31℃,固液分離;
3)用母液多次洗滌上述固體,再于25℃下自然干燥,得到深綠色立方晶體,產率約為76.7%。
實施例2
按照實施例1的方式進行,產率為74.3%,所不同的是,步驟2)中的反應溫度為65℃。
實施例3
按照實施例1的方式進行,產率為71.9%,所不同的是,步驟2)中的反應溫度為70℃。
實施例4
按照實施例1的方式進行,產率為68.9%,所不同的是,步驟1)中鐠鹽、h2l、4,4'-雙(1h-咪唑-1-取代)-1,1'-聯苯的摩爾比為12:2.0:1.2。
實施例5
按照實施例1的方式進行,產率為66.3%,所不同的是,步驟1)中鐠鹽、h2l、4,4'-雙(1h-咪唑-1-取代)-1,1'-聯苯的摩爾比為12:1.8:1.0。
實施例6
按照實施例1的方式進行,產率為47.1%,所不同的是,步驟1)中鐠鹽、h2l、4,4'-雙(1h-咪唑-1-取代)-1,1'-聯苯的摩爾比為12:1:0.8。
實施例7
按照實施例1的方式進行,產率為50.2%,所不同的是,步驟1)中鐠鹽、h2l、4,4'-雙(1h-咪唑-1-取代)-1,1'-聯苯的摩爾比為12:2.5:2。
實施例8
按照實施例1的方式進行,產率為75.2%,所不同的是,步驟1)中n,n-二甲基甲酰胺與水的混合溶液的用量為6ml。
對比例1
按照實施例1的方式進行,產率為19.4%,所不同的是,步驟2)中的反應溫度為40℃。
對比例2
按照實施例1的方式進行,產率為25.7%,所不同的是,步驟2)中的反應溫度為80℃。
對比例3
按照實施例1的方式進行,產率為23.3%,所不同的是,步驟1)中鐠鹽、h2l、4,4'-雙(1h-咪唑-1-取代)-1,1'-聯苯的摩爾比為10:0.8:0.7。
對比例4
按照實施例1的方式進行,產率為21.3%,所不同的是,步驟1)中鐠鹽、h2l、4,4'-雙(1h-咪唑-1-取代)-1,1'-聯苯的摩爾比為10:5:5。
對比例5
按照實施例1的方式進行,產率為19.6%,所不同的是,步驟1)中n,n-二甲基甲酰胺與水的混合溶液的用量為3ml。
對比例6
按照實施例1的方式進行,產率為13.7%,所不同的是,步驟1)中n,n-二甲基甲酰胺與水的混合溶液的用量為8ml。
檢測例1
在25℃下,用經過石墨單色器單色化的mokα射線(λ=0.071073nm),使用
表1
r1=σ||fo|-|fc||/|σ|fo|.wr2={σ[w(fo2-fc2)2]/σ[w(fo2)2]}1/2.
表2
對稱操作:(i)-x+1,-y+1,-z+1;(ii)-x+1,-y,-z+2;(iii)-x+2,-y+1,-z+1
通過圖1可知,配合物每個最小不對稱單元中含有一個pr(iii)離子、1.5個l2-配體、一個參與配位的dmf,兩個配位水分子和一個游離的dmf分子。pr原子采取輕微扭曲的八面體構型,每個pr(iii)離子都與八個氧原子配位,其中五個氧原子(o1、o3、o4、o5、o6)來自l2-配體,一個氧原子(o7)來自參與配位的溶劑dmf分子和兩個氧原子(o8、o9)來自配位的水分子。pr-o鍵的鍵長在
按照相同的方法對實施例2-8以及對比例1-6中的產物檢測,檢測結果與實施例1的檢測結果基本保持一致。
檢測例2
通過brukersmartapexccd單晶衍射儀對實施例1中的晶體進行檢測,檢測結果為:該配合物為三維多孔結構,脫溶劑后的孔隙率可以達到22.6%。
按照相同的方法對實施例2-8以及對比例1-6中的產物檢測,檢測結果與實施例1的檢測結果基本保持一致。
檢測例3
熱穩定測定:以5℃/min升溫速率掃描實施例1的產物的tg曲線,掃描范圍溫度范圍25-1200℃。采用dsc/tgpanal2o3熱重分析儀進行測定,檢測結果見圖3。
結果顯示:第一步在32.2-126.1℃大約失重15.54%,所對應失去的是游離的dmf分子和配位的水分子,與失重的理論值(15.89%)基本一致;第二步在126.1-341.1℃大約失重11.28%,對應失去的是配位的dmf分子,與對應的理論值(10.65%)基本一致;伴隨溫度的逐漸升高,配體發生脫落,隨即結構發生坍塌。可見該材料具有很好的熱穩定性(圖3)。
檢測例4
紫外-可見光譜性質表征:將實施例1的產物與氧化鎂混合研磨壓薄片測定,通過uv-2450,shimadzu型紫外-可見漫反射光譜儀進行紫外-可見光譜性能測試紫外-可見光譜儀的波長范圍200-800nm。具體結果見圖4(其中1曲線代表實施例1的產物,l曲線代表2,2'-聯吡啶-4,4'-二羧酸),圖5是鐠配合物的光能對應于能量的kubelka-munk函數的變形圖,通過計算得到的該晶體的能帶寬度為2.88ev,表明此材料可實現它的光催化。
檢測例5
紅外光譜性質表征:將實施例1的產物與kbr混合研磨壓薄片測定。通過irprestige-21,日本島津型號ft-ir紅外光譜儀進行測定,紅外光譜儀的波長范圍400-4000cm-1。具體結果見圖6。
由圖可知:主要紅外光譜數據(kbr壓片,cm-1):1672(s)、1652(m)、1591(m)、1544(s)、1409(s)、1261(s)、1106(s)、1058(s)、991(m)、930(m)、870(m)、775(s)、687(s)、546(m),進而說明2,2,-聯吡啶-4,4-二羧酸配體有發生紅移的現象,可判斷金屬與配體發生配位。
檢測例5
x-射線粉末衍射光譜表征:在d8-a25,bruker-axs型號x-射線粉末衍射儀上測定,x-射線粉末衍射儀的角度范圍5°-50°。具體結果見圖7,由圖可知測定的配合物的主峰均與模擬的基本相同,說明我們合成的配合物的晶體與單晶解析分析結果一致。
應用例1
1)稱取實施例1的產物20mg樣品降解30ml偶氮染料溶液(6.67mg/l),將其在暗處攪拌30min,使催化劑和染料達到吸附-解析平衡,然后用400w紫外燈光照。每隔10min,用膠頭滴管取一次樣,用離心機離心取上層清液。配合物的光催化降解性能通過紫外-可見分光光度計對其吸光光度檢測分析。具體結果見圖8和圖9。
2)按照1)中相同的方法進行,所不同的是進行單因素變量檢測,具體為的單因素變量為:將紫外燈光改為可見光、將紫外燈光改為無光、舍去催化劑。具體結果見圖8以及圖10-12。
其中,圖8是晶體的在不同條件下對偶氮染料的催化活性譜圖;圖9為晶體作為催化劑,紫外光照下的紫外-可見吸收光譜圖;圖10為晶體作為催化劑,可見光照下的紫外-可見吸收光譜圖;圖11為晶體作為催化劑,無光照下的紫外-可見吸光譜圖;圖12為無催化劑,紫外光照下的紫外-可見吸收光譜圖。
通過上圖可知,本發明提供的新型鐠配合物光催化材料在紫外線的條件下對于降解偶氮染料具有優異的光催化性能。
以上詳細描述了本發明的優選實施方式,但是,本發明并不限于上述實施方式中的具體細節,在本發明的技術構思范圍內,可以對本發明的技術方案進行多種簡單變型,這些簡單變型均屬于本發明的保護范圍。
另外需要說明的是,在上述具體實施方式中所描述的各個具體技術特征,在不矛盾的情況下,可以通過任何合適的方式進行組合,為了避免不必要的重復,本發明對各種可能的組合方式不再另行說明。
此外,本發明的各種不同的實施方式之間也可以進行任意組合,只要其不違背本發明的思想,其同樣應當視為本發明所公開的內容。
- 還沒有人留言評論。精彩留言會獲得點贊!