人類親權鑒定用多重PCR引物組及檢測方法與流程
本發明涉及醫療檢測領域,通過使用特定的snp標記組合對人類親權關系進行檢測,具體涉及表一中部分或全部核苷酸序列以及檢測方法。
背景技術:
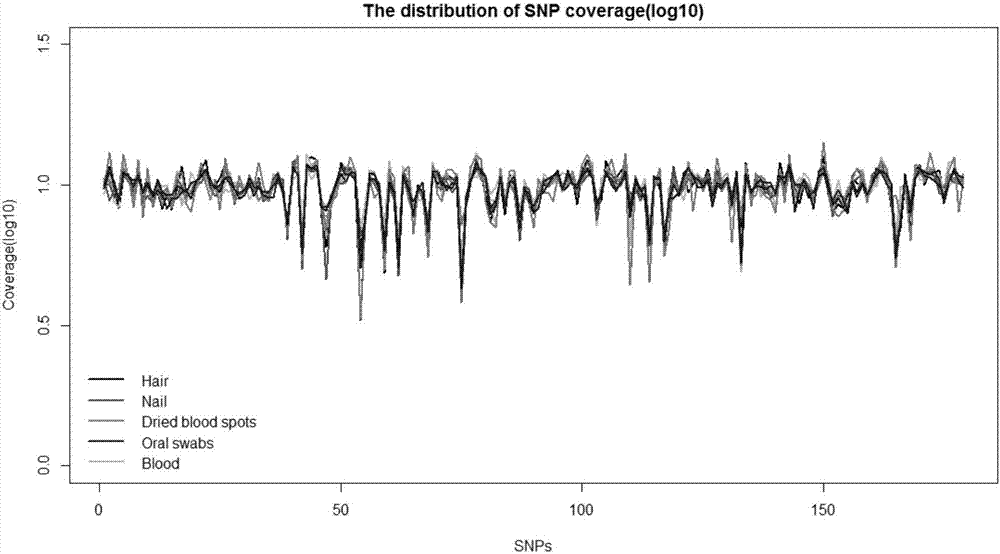
:親權鑒定是指通過對人類遺傳標記的檢測,根據遺傳規律分析,對個體之間血緣關系的鑒定。目前用于親權鑒定的方法是dna分析,由于最早時的第一代遺傳標記--限制性片段長度多態性(rflp)具有自身的局限性,檢測過程費時費力,且一次僅能檢測一個位點,多態性程度較低,因此在親權鑒定中的應用逐漸減少;此后以第二代遺傳標記--短串聯重復序列(str)為主的分型技術成為個體識別與親權鑒定研究中使用頻率最高的分子標記,它具有信息量大、分布廣泛、易于檢測、多態性程度高且遵循孟德爾共顯性遺傳等特點。在應用str進行親權鑒定時,檢測的基因座需要達到15-20個,若出現3個及3個以上違反遺傳規律的基因座,則可排除親權關系。然而,近年來發現str標記判型錯誤率較高,重復性差,會直接影響親權鑒定的準確性和可重復性。單核苷酸多態性(snp)作為繼rflp和str后的第三代遺傳標記,越來越多地受到人們的關注。目前大量的snp標記已經被應用于基因功能研究和疾病的關聯分析中,并已逐漸取代str,成為首選的遺傳標記,snp標記因其具有分布廣泛,數量豐富,遺傳穩定,高通量檢測快速準確等其他標記無法比擬的優勢,因此在無創親子鑒定領域中成為新興熱門的鑒定方法。起初,孕婦懷孕期間,但又不確定懷孕的胎兒父親是誰,能解決的方法是在懷孕期間進行絨毛膜取樣或羊膜穿刺,但這會帶來流產的風險,另外的一種方法是等胎兒出生后再去鑒定生父。直到1997年,lo等人研究證實,孕婦的血液中存在胎兒游離dna,這為無創親子鑒定提供了技術依據。隨著高通量測序技術的發展,基于snp標記位點對在懷孕期間對胎兒進行無創親子鑒定成為親權鑒定的主要發展方向。但用于無創鑒定的胎兒游離dna數量有限,snp標記分布眾多,富集過程容易導致測序結果讀取(reads)數目分布差異較大,背景噪音引起結果偏差,基因組突變差異對snp遺傳差異造成較大干擾,容易造成懷孕早期無創檢測結果的準確性降低。技術實現要素:有鑒于此,本發明創造旨在提出一組人類親權鑒定用多重pcr引物組及檢測方法,能夠克服懷孕早期無創親子鑒定的不足,準確度高,可應用于司法鑒定以及法醫學研究中。本發明創造提供的人類親權鑒定用多重pcr引物組,所涉及的snp位點及引物滿足以下條件:t1:常染色體上snp位點的maf值大于0.4;t2:x染色體上snp位點的maf值大于0.3;t3:y染色體上snp位點的maf值大于0.1;t4:maf的頻率在北京人種和亞洲人種中同時要滿足上述t1,t2,t3頻率要求;t5:所選的snp位點不能位于人類常見cnv區域;t6:所選snp位點與其相鄰3bp組成的區域不能是3個或3個以上相同的堿基重復序列;t7:所選snp位點與其左右50bp組成的區域不能存在循環(repeat)序列;t8:所選snp位點上已知的基因型不能超過兩種;t9:所選snp位點之間的距離要大于4,000,000bp;t10:對snp左右75bp進行發夾結構,二聚體的預測,去除存在發夾結構或二聚體的snp位點;t11:把snp左右50bp的序列切割成30bpk-mer,重新比對到基因組,比對質量相加,最大者優先選擇;t12:對上述所選snp位點進行高通量測序,進行重復測序實驗(優選為重復10組實驗),計算每個snp位點測得的reads數目中位數,計算這些中位數的標準差,去除中位數大于或小于標準差3倍的snp位點。其中,所述多重pcr引物組中所涉及的snp位點不少于150個,優選地不少于179個,更優選地不少于400個。根據上述條件,篩選并建立了一組由snp遺傳標記位點組成的多重pcr引物組,具體見后面表一所示。其中,所述多重pcr引物組優選地由表一中的任意150對以上的引物組構成,更優選地由表一中的任意179對以上的引物組構成,還優選地由表一中的任意400對以上的引物組構成,進一步優選地由表一中的前179對以上的引物組構成,最優選地由表一中的前415對以上的引物組構成。本發明還提供了采用上述多重pcr引物組進行人類親權鑒定的檢測方法,包括下述步驟:s1:提取孕婦新鮮外周血漿中的胎兒游離dna;提取基因組dna,包括孕婦dna、疑似父親dna、隨機未知父親(例如,可以為亞洲任一隨機男子)dna;s2:利用上述多重pcr引物組進行多重pcr擴增,分別對隨機未知父親dna、疑似父親dna、孕婦dna和胎兒游離dna進行富集;s3:分別對擴增后的pcr產物進行文庫構建、稀釋后,采用高通量測序方法進行測序;s4:根據測序結果對疑似父親、孕婦、胎兒的snp位點對應的基因型進行比對,進行親權關系推斷。其中,所述疑似父親dna可以包括從外周血中提取的gdna、從干血片中提取的gdna、從口腔上皮細胞中提取的gdna、從毛發(帶毛囊)中提取的gdna、或從指甲中提取的gdna。其中,所述步驟s3中,高通量測序可采用新一代高通量測序儀,如ionproton平臺或illumina平臺進行上機測序。其中,所述步驟s4中,所述親權關系推斷步驟包括:s4-1:選取測序深度大于200的snp位點;s4-2:選取孕婦snp位點中最大等位基因reads頻率大于99%的位點;s4-3:計算父親指數paternityindex(pi)和累計親權指數combinedpaternityindex(cpi),pi=x/yx=pr(plasma|mother,t)代表嫌疑父親為胎兒生物學父親的概率;y=pr(plasma|mother,r)隨機未知父親為胎兒生物學父親的概率;根據貝葉斯模型:一般當cpi值大于10000時,我們認為檢測的疑似父親和孕婦所懷的胎兒有生物學上親子關系。相對于現有技術,本發明創具有以下優勢:本發明通過高通量測序技術檢測snp多態性并對親權關系進行推斷及驗證,無論是二聯體親子鑒定還是三聯體親子鑒定,其親權關系都是極為顯著的,并且實驗鑒定結果與實際親權關系相吻合;本發明提供的多重pcr引物組的篩選條件,以及根據該條件確認的由表一中部分或全部snp遺傳標記位點組成的多重pcr引物組,能夠在懷孕10周即可進行無創檢測,同時能夠消除富集過程導致的測序結果reads數目分布差異,降低背景噪音,減少基因組突變干擾,有效提高懷孕早期無創檢測結果的準確性,當多重pcr引物組中所涉及的snp位點不少于150個時,基本能夠保證十萬分之一的檢測精度。附圖說明圖1為本發明多重pcr引物組進行無創親子鑒定檢測的不同樣本類型分析圖。具體實施方式下面結合具體實施例來詳細說明本發明創造。但不用于限定本發明。本文中,“以上”的表述包括本數。一、采用表一所示的部分或全部snp遺傳標記位點組成的多重pcr引物組進行人類親權檢測,以下為具體步驟。a.提取dna本實驗的孕婦樣本是采用ardent采血管采集的全血,通過兩步離心法獲得血漿,具體操作如下:4℃,1600g,離心10min,吸取上清到新的2mlep管中。收集到的上清液再在4℃條件下,16000g,離心10min,同樣吸取上清到新的2mlep管中,備用。(1)提取胎兒游離dna1)取2ml血漿,平衡至室溫,加入30ulproteinasek(濃度為20mg/ml),100ul20%的sds溶液,振蕩混勻后,短暫離心,于60℃孵育20min。反應完成后,冰上孵育5min。2)將30ulcellfreednamagneticbeads加入到2.5ml的cellfreednalysis/bindingsolution中,振蕩混勻,配制成混合液,加入到第一步反應液中,充分渦旋混勻,使用垂直混勻儀搖勻10min。3)取下離心管,將其置于磁力架上,靜置5min,待溶液澄清后棄上清。4)取下離心管,加入1mlcellfreednawashsolution,重懸磁珠,短暫離心后置于磁力架上,2min后溶液澄清,棄上清。5)重復步驟4)一次。6)取下離心管,加入1ml當天配制的80%酒精,重懸磁珠,短暫離心后置于磁力架上,2min后溶液澄清,棄上清。7)重復步驟6)一次。8)繼續將離心管置于磁力架上,開蓋晾干10min。9)待磁珠晾干后,加入25ulcellfreednaelutionsolution,渦旋混勻5min,短暫離心。10)再次將離心管放置于磁力架上,2min后溶液澄清,轉移上清液到新的1.5mlep管中,并做好標記。11)使用qubit2.0定量cfdna濃度后可立即做后續實驗或存放于-20℃保存。(2)提取孕婦gdna1)取1.5mlep管,依次加入20ulqiagenprotease、200ul全血,200ulbufferal,渦旋混勻15s。2)將ep管于56℃,孵育10min。3)短暫離心后加入200ul乙醇(96-100%)至上述反應液中,再次渦旋混勻15s,短暫離心。4)轉移反應液于吸附柱中,8000rpm離心1min,棄掉廢液,將吸附柱放置于干凈的2ml收集管中。5)加入500ulbufferaw1于吸附柱中,8000rpm離心1min,棄掉廢液,將吸附柱放置于干凈的2ml收集管中。6)加入500ulbufferaw2于吸附柱中,14000rpm離心3min,棄掉廢液,將吸附柱放置于干凈的2mlep管中,再次14000rpm離心1min。7)取下吸附柱,放置于干凈的1.5mlep管中,加入200ulbufferae,室溫孵育5min。8)8000rpm離心1min,收集到的溶液即為最終的gdna。9)使用nanodrop和qubit2.0定量gdna濃度后可立即做后續實驗或存放于-20℃保存。(3)提取疑似父親gdna(口腔拭子,隨機未知父親的相關dna提取方法相同)1)將口腔拭子取出放入2ml離心管中,依次加入400ulpbs、20ulqiagenprotease、400ulbufferal,渦旋混勻15s。2)將離心管于56℃,孵育10min。3)短暫離心后加入400ul乙醇(96-100%)至上述反應液中,再次渦旋混勻15s,短暫離心。4)轉移反應液于吸附柱中,8000rpm離心1min,棄掉廢液,將吸附柱放置于干凈的2ml收集管中。5)重復步驟4)一次。6)加入500ulbufferaw1于吸附柱中,8000rpm離心1min,棄掉廢液,將吸附柱放置于干凈的2ml收集管中。7)加入500ulbufferaw2于吸附柱中,14000rpm離心3min,棄掉廢液,將吸附柱放置于干凈的2mlep管中,再次14000rpm離心1min。8)取下吸附柱,放置于干凈的1.5mlep管中,加入150ulbufferae,室溫孵育5min。9)8000rpm離心1min,收集到的溶液即為最終的gdna。10)使用nanodrop和qubit2.0定量gdna濃度后可立即做后續實驗或存放于-20℃保存。(4)提取疑似父親gdna(干血片,隨機未知父親的相關dna提取方法相同)1)用打孔器從干血片中切出3~5片直徑為3mm的帶血圓片,并轉移至1.5ml離心管中。2)依次加入200ulbufferatl、20ulproteinasek,于55℃振蕩孵育1h。3)加入220ulbufferal至上述反應液中,于70℃振蕩孵育15min。4)10000g,離心3min,收集管中液體,并將其轉移至新的干凈的1.5ml離心管中。5)依次加入20ulmagbindparticles、400ulbuffermbd,振蕩混勻6min。6)短暫離心,將離心管置于磁力架,2min后溶液澄清,棄上清。7)加入500ulbuffergw1,振蕩混勻1min。8)短暫離心,將離心管置于磁力架,2min后溶液澄清,棄上清。9)重復步驟7)~8)一次。10)加入500ul75%乙醇,振蕩混勻1min。11)短暫離心,將離心管置于磁力架,2min后溶液澄清,棄上清。12)重復步驟10)~11)一次。13)繼續將離心管置于磁力架上,晾干磁珠6min。14)加入30ulbufferae于上述晾干的磁珠中,于60℃振蕩孵育10min。15)短暫離心,將離心管置于磁力架,2min后溶液澄清,收集上清液并轉移至干凈的1.5ml離心管中。16)使用nanodrop和qubit2.0定量gdna濃度后可立即做后續實驗或存放于-20℃保存。(5)提取疑似父親dna(其他,隨機未知父親的相關dna提取方法相同)其他疑似父親dna樣本可采用常規方法獲得。為驗證疑似父親dna樣本不同來源對檢測結果的影響,分析了上述表一中全部snp遺傳標記位點分別在不同來源dna樣本中的分布情況(圖1),可以看出,不同dna樣本來源中,上述snp覆蓋率的分布基本相同,確保了不同樣本來源檢測結果的一致性和準確性。b.多重pcr擴增1)將多重pcr引物panel、gdna于冰上解凍,備用。2)多重pcr擴增的反應體系如下:componentvolume(ul)5xionampliseqtmhifimix4ulprimerpool10ul模板dna(5~18ng)≤6ulnuclease-freewaterto20ul槍頭吹打混勻后,短暫離心,將pcr反應管置于pcr儀上反應,反應程序如下:反應結束后,取出pcr管短暫離心。c.文庫構建采用文庫構建試劑盒對擴增后的多重pcr產物進行文庫構建。1)部分消化引物序列加入2ulfupareagent到每個樣本中,此時總體積為22ul。槍頭吹打混勻后,短暫離心,將pcr反應管置于pcr儀上反應,反應程序如下:反應結束后,取出pcr管短暫離心。2)連接接頭首先按照p1adapter:barcodex:nuclease-freewater=1:1:2的比例稀釋接頭。之后按照如下反應體系依次加入每個樣本中:componentvolume(ul)switchsolution4ul稀釋好的接頭2uldnaligase2ultotal30ul槍頭吹打混勻后,短暫離心,將pcr反應管置于pcr儀上反應,反應程序如下:temperaturetime22℃30min72℃10min10℃hold(最多1小時)反應結束后,取出pcr管短暫離心。3)純化上述dna片段dna純化磁珠在使用前需平衡30min至1h。①吸取dna純化磁珠,吸取的量為1.5~3倍樣本體積,用同一槍頭吹打混勻,室溫靜置5min。②將pcr管放在96孔磁力架上,5min后溶液變澄清,棄去上清液。③吸取150ul當天配制的70~75%乙醇于上述反應磁珠中;在磁力架中來回移動pcr管的位置使磁珠得到充分洗脫,棄去上清液。④重復步驟③一次。⑤盡量吸去殘留溶液后,將磁珠暴露空氣中,室溫晾干磁珠5min。⑥配制pcr反應混合液,反應體系如下:⑦吸取配制好的反應混合液52ul于上步晾干的磁珠中,用同一槍頭吹打混勻后,靜置反應5min。⑧將pcr管置于磁力架上,待溶液澄清后將全部上清液(約52ul)吸取至新的pcr管中,pcr反應程序如下:反應結束后,取出pcr管短暫離心。4)文庫擴增后的片段篩選及純化①吸取dna純化磁珠,吸取量為0.5~2倍樣本體積,用同一槍頭吹打混勻,室溫靜置5min。②將pcr管放在96孔磁力架上,5min后溶液變澄清,轉移上清液至新的pcr管中。③吸取dna純化磁珠,吸取量為1.2~2.5倍樣本體積,加到上清液中,用同一槍頭吹打混勻,室溫靜置5min。④將pcr管放在96孔磁力架上,5min后溶液變澄清,棄去上清液。⑤吸取150ul當天配制的70~75%乙醇于上述反應磁珠中;在磁力架中來回移動pcr管的位置使磁珠得到充分洗脫,棄去上清液。⑥重復步驟⑤一次。⑦盡量吸去殘留溶液后,將磁珠暴露空氣中,室溫晾干磁珠5min。⑧取下pcr管,并加入50ullowte至晾干的磁珠中,用槍頭吹打混勻,室溫放置5min。⑨重新將pcr管放回96孔磁力架上,溶液澄清后,轉移上清50ul到干凈的1.5mlep管中,做好標記。⑩制備好的文庫可存放于-20℃保存。d.文庫質控采用qubit2.0和agilent2100對構建好的文庫進行質控,并將文庫稀釋至100~200pm之間,準備上機測序。e.測序應用新一代高通量測序儀,如ionproton平臺或illumina平臺進行上機測序。f.數據分析通過軟件對上機測序的結果進行生物信息學分析,對疑似父親、孕婦以及胎兒的snp位點對應的基因型進行比對,通過計算親權指數,進一步得到父權相對機會,以此進行親權關系推斷。二、檢測結果的驗證為了驗證所篩選的snp遺傳標記位點組成的多重pcr引物組在實際家庭中進行親權鑒定的可行性和準確性,本實施例從中山開發區醫院采集了20組樣本(孕婦孕周均為10周-12周)進行無創親子鑒定的驗證實驗,其中有10組是夫妻關系明確的家庭組合,剩余10組為無關未知男性與任一孕婦配對組合。將這20組樣本分別編號為ky-family1~ky-family20。通過實施例1中的檢測方法對20組樣本進行親緣關系的推斷,179個snp遺傳標記位點組成的多重pcr引物組(表一中的前179對引物組)和415個snp遺傳標記位點組成的多重pcr引物組(表一中的前415對引物組)相比較的鑒定結果見表二。表二注:父權相對機會(rcp):由于親權指數是實數,由數字不易看出父權的機會,習慣上將父權指數換算成一個條件概率,即父權相對機會,也是親子關系概率,代表了判斷疑似父親是孩子生父的把握度大小。由表二可以看出,本發明提供的篩選條件及其篩選出的多重pcr引物組用于無創親子鑒定時,其親權關系顯著,179個snp遺傳標記位點組成的多重pcr引物組鑒定親權關系時其父權相對機會可達99.999%以上,能夠對親權關系作出準確地區分。但增加位點數至415時,父權相對機會能夠達到99.999999%以上,且多位點分析時結果更接近真實情況,因此,本發明優選地采用415位點組成的多重pcr引物panel進行無創親子鑒定,結果準確性高,鑒定結果與實際親權關系完全吻合;利用新一代高通量測序技術檢測snp多態性,以此進行無創產前親子鑒定,具有安全、快速、準確、低成本、高通量等諸多優勢,并且相比于目前已有的檢測方法,該發明在孕婦懷孕的早期孕周10周時即可進行檢測,且胎兒濃度可達到6%以上,除此之外,整個檢測過程只需采集孕媽外周血,安全無創傷,為更多的家庭或個人提供巨大的便利和價值,同時也可將該方法應用于法學研究的親權鑒定中,具有很大的應用前景和經濟效益。表一以上所述僅為本發明創造的較佳實施例而已,并不用以限制本發明創造,凡在本發明創造的精神和原則之內,所作的任何修改、等同替換、改進等,均應包含在本發明創造的保護范圍之內。當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1