一種可回收溶劑中制備吡唑類化合物的方法與流程
本發明屬于醫藥化工中間體及相關化學技術領域,涉及到可回收溶劑體系二甲基亞砜(dmso)/h2o在制備吡唑類化合物中的應用。
背景技術:
吡唑類化合物在多個領域有著非常廣泛的應用,尤其在醫藥和農藥方面表現出優異的性能。吡唑類化合物作為除草劑和殺蟲劑,具有作用廣譜、藥效強、低毒、低殘留等優點;作為醫藥分子,對癌癥、哮喘、關節炎等疾病具有較好的療效。此外,吡唑類化合物可用于造紙、皮革、洗滌、塑料、涂料等行業。
傳統的合成吡唑類化合物的方法主要包括(1)由重氮化物與烯烴或炔烴合成吡唑;(2)由肼亞胺與烯烴或炔烴合成吡唑;(3)由肼與1,3-二羰基化合物合成吡唑;(4)由肼與烯酮合成吡唑;(5)由肼與炔酮合成吡唑;(6)由肼與炔烴合成吡唑。這些合成方法均使用毒性較大或難以回收的溶劑作為反應媒介,在溶劑回收使用中存在對環境的污染大、回收成本高等問題。因而,開發一種毒性小、易回收使用的制備吡唑類化合物的溶劑,可降低生產成本和對人類健康的危害,對推動我國環保事業的發展,具有重要的意義。
技術實現要素:
本發明為解決現有技術問題,提供了一種使用低毒、廉價、易回收的溶劑制備吡唑類化合物的方法。
本發明采用技術方案為:一種可回收溶劑中制備吡唑類化合物的方法,制備原料為1,3-二炔
其具體制備方法包含以下步驟:(a)先將1,3-二炔加入到schlenk瓶中;在氮氣保護下將85%的肼水溶液和dmso/h2o溶劑加入到schlenk瓶中;(b)將schlenk瓶置于油浴中進行反應,反應結束后冷卻至室溫得到反應液;(c)對反應液進行萃取、靜置分層,下層得到反應溶劑上層得到目標溶液;(d)對目標溶液依次進行無水硫酸鈉干燥、減壓蒸餾、洗脫劑洗脫后即得到白色固體產物。
進一步地,所述1,3-二炔上的r1或r2可以是苯環、吡啶環、噻吩環、環己環、烷基鏈中的任意一種基團。
進一步地,所述肼與1,3-二炔的摩爾比為1:1~6:1。
進一步地,所述溶劑dmso/h2o中dmso與h2o的體積比為1:1~5:1,優選為2:1~3:1。
進一步地,所述步驟(c)中萃取所得的反應溶劑dmso/h2o可直接再次使用。
進一步地,所述油浴的反應溫度為80~150℃,反應時間為12~36h。
進一步地,所述萃取劑為乙酸乙酯、石油醚、環己烷、二氯甲烷或體積比為1:5~5:1的乙酸乙酯/石油醚,優選體積比為1:1~3:1的乙酸乙酯/石油醚。
進一步地,所述萃取劑與反應溶劑的體積比為1:2~5:1,優選1:1~2:1。
進一步地,所述肼與1,3-二炔的摩爾比為3:1~4:1。
進一步地,所述油浴的反應溫度為100~120℃,反應時間為15~20h。
本發明獲得的有益效果為:所述的吡唑類化合物的制備方法,反應步驟較少,原料價格低廉,溶劑易回收使用、反應條件溫和,環境友好;并且所得產品收率高、純度高、完全符合作為藥物中間體的質量要求,為其工業化生產提供了有利途徑,展現出良好的應用前景。
附圖說明
圖1為本發明制備方法的操作路線圖
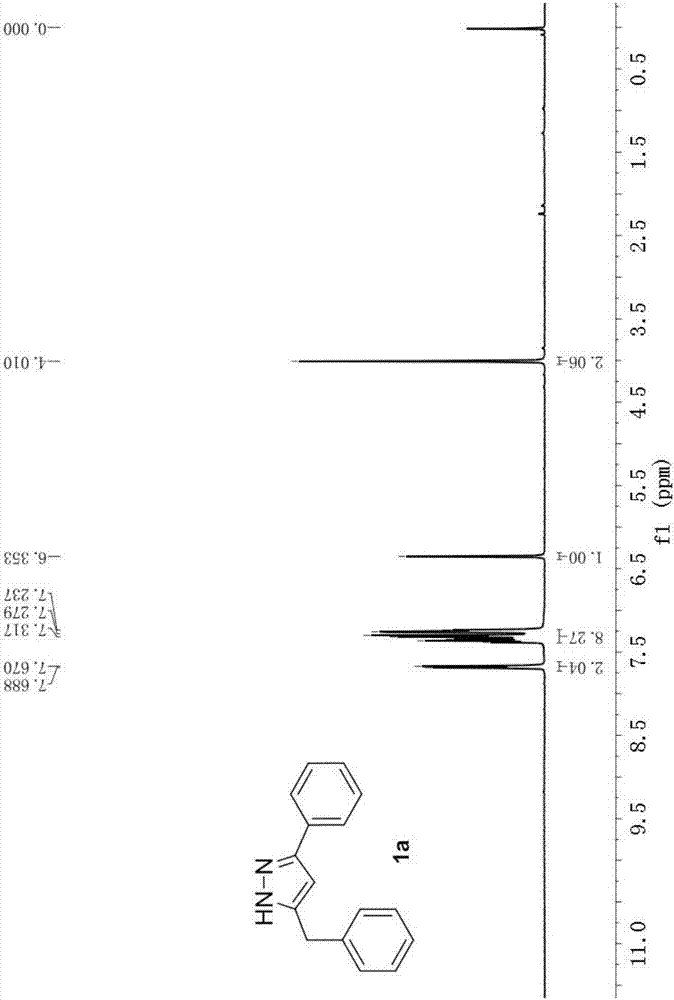
圖2為實施例1中化合物1a的1h-nmr
圖3為實施例1中化合物1a的13c-nmr
圖4為實施例2中化合物1b的1h-nmr
圖5為實施例2中化合物1b的13c-nmr
圖6為實施例3中化合物1c的1h-nmr
圖7為實施例3中化合物1c的13c-nmr
圖8為實施例4中化合物1d的1h-nmr
圖9為實施例4中化合物1d的13c-nmr
圖10為實施例5中化合物1e的1h-nmr
圖11為實施例5中化合物1e的13c-nmr
圖12為實施例6中化合物1f的1h-nmr
圖13為實施例6中化合物1f的13c-nmr
具體實施方式
下面結合具體實施例,進一步闡述本發明,這些實施例僅用于說明本發明而不用于限制本發明的范圍,在本領域內的技術人員對本發明所做的簡單替換或改進均屬于本發明所保護的技術方案之內。
實施例1:5-benzyl-3-phenyl-1h-pyrazole(1a)的制備,具體實施如下:
(a)準確稱取1,4-二苯基丁二炔(101.1mg,0.5mmol),加入到25ml的schlenk瓶中。(b)將schlenk瓶使用氮氣置換3次后,在氮氣保護下加入85%的肼水溶液(75.3μl,2.0mmol),dmso/h2o(1:1,4ml),然后置于90℃的油浴中反應15h。(c)反應結束后,使用3×4ml乙酸乙酯萃取3次,合并有機相,使用無水硫酸鈉干燥20min后減壓除去溶劑,使用石油醚/乙酸乙酯作為洗脫劑,硅膠柱分離,得到白色固體產物5-benzyl-3-phenyl-1h-pyrazole(1a)(收率為84%,98.4mg)。1hnmr(400mhz,cdcl3)δ7.68(d,j=7.2hz,2h),7.38–7.24(m,8h),6.35(s,1h),4.01(s,2h);13cnmr(100mhz,cdcl3)δ138.6,132.1,128.9,128.8,128.1,126.8,125.8,102.2,33.3.
實施例2:5-(4-fluorobenzyl)-3-(4-fluorophenyl)-1h-pyrazole(1b)的制備,具體實施如下:
(a)準確稱取1,4-二(4-氟苯基)丁二炔(119.1mg,0.5mmol),加入到25ml的schlenk瓶中。(b)將schlenk瓶使用氮氣置換3次后,在氮氣保護下加入85%的肼水溶液(56.5μl,1.5mmol),dmso/h2o(2:1,4ml),然后置于100℃的油浴中反應16h。(c)反應結束后,使用3×5ml乙酸乙酯/石油醚(2:1)萃取3次,合并有機相,使用無水硫酸鈉干燥20min后減壓除去溶劑,使用石油醚/乙酸乙酯作為洗脫劑,硅膠柱分離,得到白色固體產物5-(4-fluorobenzyl)-3-(4-fluorophenyl)-1h-pyrazole(1b)(收率為92%,124.3mg)。1hnmr(400mhz,cdcl3)δ9.56(s,1h),7.59(dd,j=8.0,5.6hz,2h),7.12(dd,j=8.0,5.6hz,2h),7.00(dd,j=8.4,8.4hz,2h),6.95(dd,j=8.4,8.4hz,2h),6.25(s,1h),3.90(s,2h);13cnmr(100mhz,cdcl3)δ162.8(d,1j=245.7hz),161.9(d,1j=243.5hz),148.9,147.2,134.1,130.2(d,3j=7.9hz),128.4,127.5(d,3j=8.1hz),115.8(d,2j=20.8hz),115.6(d,2j=20.5hz),102.1,32.3.
實施例3:5-(naphthalen-1-ylmethyl)-3-(naphthalen-2-yl)-1h-pyrazole(1c)的制備,具體實施如下:
(a)準確稱取1,4-二(2-萘基)丁二炔(151.2mg,0.5mmol),加入到25ml的schlenk瓶中。(b)將schlenk瓶使用氮氣置換3次后,在氮氣保護下加入85%的肼水溶液(37.7μl,1.0mmol),dmso/h2o(1:1,5ml),然后置于110℃的油浴中反應20h。(c)反應結束后,使用3×5ml乙酸乙酯/石油醚(3:1)萃取3次,合并有機相,使用無水硫酸鈉干燥20min后減壓除去溶劑,使用石油醚/乙酸乙酯作為洗脫劑,硅膠柱分離,得到白色固體產物5-(naphthalen-1-ylmethyl)-3-(naphthalen-2-yl)-1h-pyrazole(1c)(收率為71%,118.7mg)。1hnmr(400mhz,cdcl3)δ10.68(s,1h),8.22(d,j=8.0hz,1h),7.89(d,j=7.6hz,1h),7.80–7.64(m,4h),7.44–7.18(m,8h),6.18(s,1h),4.18(s,2h);13cnmr(100mhz,cdcl3)δ147.4,134.8,134.0,133.9,132.0,131.4,130.0,128.8,128.7,128.4,127.5,127.2,126.9,126.5,126.2,126.0,125.82,125.75,125.7,125.3,124.1,30.7.
實施例4:3-cyclohexyl-5-(cyclohexylmethyl)-1h-pyrazole(1d)的制備,具體實施如下:
(a)準確稱取1,4-二環己基丁二炔(107.2mg,0.5mmol),加入到25ml的schlenk瓶中。(b)將schlenk瓶使用氮氣置換3次后,在氮氣保護下加入85%的肼水溶液(75.4μl,2.0mmol),dmso/h2o(3:1,5ml),然后置于120℃的油浴中反應20h。(c)反應結束后,使用3×5ml乙酸乙酯/石油醚(1:1)萃取3次,合并有機相,使用無水硫酸鈉干燥20min后減壓除去溶劑,使用石油醚/乙酸乙酯作為洗脫劑,硅膠柱分離,得到白色固體產物3-cyclohexyl-5-(cyclohexylmethyl)-1h-pyrazole(1d)(收率為87%,107.2mg)。1hnmr(400mhz,cdcl3)δ8.54(s,1h),5.81(s,1h),2.63–2.59(m,1h),2.47(d,j=7.2hz,2h),1.99–0.88(m,21h);13cnmr(100mhz,cdcl3)δ154.2,147.7,101.1,38.4,36.6,35.2,33.4,33.2,26.6,26.42,26.36,26.2;
實施例5:3-(thiophen-3-yl)-5-(thiophen-3-ylmethyl)-1h-pyra-zole(1e)的制備,具體實施如下:
(a)準確稱取1,4-二(3-噻吩)丁二炔(107.2mg,0.5mmol),加入到25ml的schlenk瓶中。(b)將schlenk瓶使用氮氣置換3次后,在氮氣保護下加入85%的肼水溶液(113.1μl,3.0mmol),dmso/h2o(4:1,4ml),然后置于100℃的油浴中反應12h。(c)反應結束后,使用3×4ml乙酸乙酯/石油醚(5:1)萃取3次,合并有機相,使用無水硫酸鈉干燥20min后減壓除去溶劑,使用石油醚/乙酸乙酯作為洗脫劑,硅膠柱分離,得到白色固體產物3-(thiophen-3-yl)-5-(thiophen-3-ylmethyl)-1h-pyrazole(1e)(收率為82%,101.0mg)。1hnmr(400mhz,cdcl3)δ9.44(s,1h),7.44(d,j=1.6hz,1h),7.34–7.23(m,3h),6.96–6.91(m,2h),6.24(s,1h),3.94(s,2h);13cnmr(100mhz,cdcl3)δ147.0,145.0,138.8,133.5,128.4,126.3,126.0,121.8,121.0,102.2,27.8.
實施例6:5-(4-nitrobenzyl)-3-phenyl-1h-pyrazole(1f)的制備,具體實施如下:
(a)準確稱取1-4硝基苯基-4-苯基丁二炔(123.6mg,0.5mmol),加入到25ml的schlenk瓶中。(b)將schlenk瓶使用氮氣置換3次后,在氮氣保護下加入85%的肼水溶液(94.2μl,2.5mmol),dmso/h2o(3:1,5ml),然后置于120℃的油浴中反應16h。(c)反應結束后,使用3×5ml乙酸乙酯/石油醚(3:1)萃取3次,合并有機相,使用無水硫酸鈉干燥20min后減壓除去溶劑,使用石油醚/乙酸乙酯作為洗脫劑,硅膠柱分離,得到白色固體產物5-(4-nitrobenzyl)-3-phenyl-1h-pyrazole(1f)(收率為93%,129.9mg)。1hnmr(400mhz,cdcl3)δ11.58(s,1h),8.08(d,j=8.4hz,2h),7.57(d,j=6.4hz,2h),7.37–7.30(m,5h),6.31(s,1h),4.03(s,2h);13cnmr(100mhz,cdcl3)δ146.8,146.7,130.7,129.6,129.1,128.6,125.7,123.9,102.5,33.7.
溶劑dmso/h2o(體積比3:1)的循環使用實驗,具體實施如下:
(a)準確稱取1,4-二苯基丁二炔(101.1mg,0.5mmol),加入到25ml的schlenk瓶中。(b)將schlenk瓶使用氮氣置換3次后,在氮氣保護下加入85%的肼水溶液(75.3μl,2.0mmol),dmso/h2o(3:1,4ml),然后置于110℃的油浴中反應15h。(c)反應結束后,使用3×4ml乙酸乙酯/石油醚(2:1)萃取3次,合并有機相,使用無水硫酸鈉干燥20min后減壓除去溶劑,使用石油醚/乙酸乙酯作為洗脫劑,硅膠柱分離,得到白色固體產物5-benzyl-3-phenyl-1h-pyrazole(1a).(d)將萃取后的dmso/h2o作為反應溶劑,重復上述(a)~(d)的操作過程。
溶劑循環使用實驗見表1:
表1溶劑dmso/h2o(3:1)的循環使用
溶劑連續循環使用第5次時,產物1a的分離收率仍為70%,溶劑的回收率為94%。溶劑連續循環第6次時,產物1a的收率為56%,溶劑的回收率為92%。當將0.5mldmso補加到第6次循環試驗的dmso/h2o中后,產物1a的收率升高到80%。
可見本發明的溶劑循環使用效果好,可作為一種合成吡唑類化合物的循環溶劑。
- 還沒有人留言評論。精彩留言會獲得點贊!