一種2?乙烯基?2,4,4,6,6?五甲基環三硅氧烷的環保制備方法與流程
本發明涉及一種2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷的環保制備方法,屬于有機硅化合物的合成領域。
背景技術:
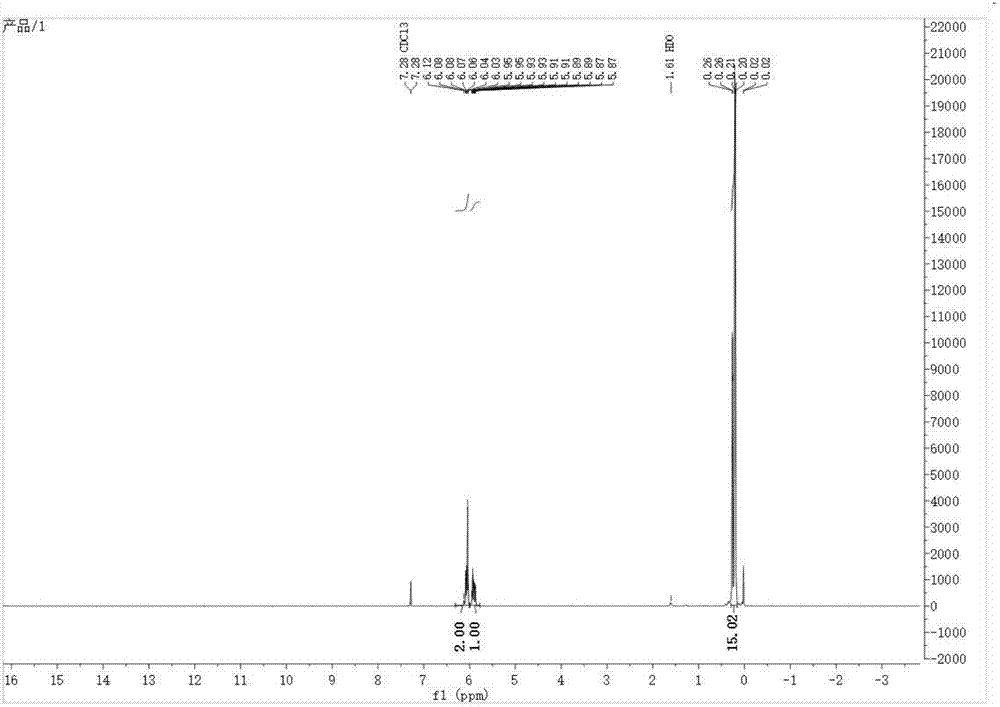
:環硅氧烷是有機硅產業中非常重要的中間體原料,被廣泛應用于合成硅油、硅樹脂和硅橡膠等。以其制備的有機硅材料具有優異的電氣性能、生理惰性、耐腐蝕、耐高低溫、耐老化等性能,是其他的有機高分子材料所不能比擬和替代的,在航空航天、電子電氣、汽車船舶、輕工紡織、建筑、機械、醫學、交通運輸等方面得到廣泛的應用,日益成為國民經濟中重要而不可缺少的新型高分子材料。2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷是將六甲基環三硅氧烷中的一個甲基取代為乙烯基的特殊有機硅環體。由于結構中只含有一個乙烯基,具有許多獨特的功能。研究顯示,在硫化體系中,分子鏈內若兩個乙烯基相鄰或幾個乙烯基擠在一起,即使硫化完全,其彈性也相當于只有一個交聯點來提供。2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷與其他有機硅環體開環聚合后,分子鏈內的乙烯基甲基硅氧鏈節處于兩個二甲基硅氧鏈節之間,避免了乙烯基聚集狀態的出現,在硫化的過程中,提高了乙烯基的交聯活性,相比于其他的乙烯基環體,乙烯基的利用率更高,固化后的效果更加顯著。目前文獻報道了甲基乙烯基硅烷混合環體的制備,以乙烯基甲基二氯硅烷單體和二甲基二氯硅烷單體為原料,酸性、低溫條件下混合水解制備了甲基乙烯基硅烷混合環體,通過氣相色譜-質譜聯用儀對有機硅甲基、乙烯基環體進行了定性分析,產品中主要包括:d3、d4、d5、d6、v1d3、v2d2、v3d1等混合環體(參見胡文斌等,.乙烯基氯硅烷和甲基氯硅烷混合水解機理研究.《化工新型材料》.2007,第35卷(第5期),第30-32頁.)。該方法采用共水解的反應機理,水解過程難以掌控,產品中環體種類復雜,無法有效制備乙烯基五甲基環三硅氧烷,乙烯基五甲基環三硅氧烷收率非常低,并且原料乙烯基氯硅烷和甲基氯硅烷穩定性很差,操作過程中,十分容易產生鹽酸汽霧,嚴重危害身體健康及環境安全。另外,現有技術中還有通過四甲基二硅氧烷二醇和乙烯基甲基二氯硅烷,在乙醚溶劑中,于三乙胺和4-二甲氨基吡啶催化下,脫除氯化氫,環化得到乙烯基五甲基環三硅氧烷。該方法使用不利環保的二氯硅烷,且在反應過程中引入較多的溶劑乙醚,同時產生大量的副產物三乙胺鹽酸鹽,對環保不利,并且制備步驟復雜,該種方法中的原料四甲基二硅氧烷二醇需通過價格昂貴的鈀/碳催化劑反應制得,成本過高。技術實現要素:針對現有技術存在的不足,本發明提供一種低成本的2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷的環保制備方法。本發明解決了現有技術中使用氯硅烷帶來的環保問題,避免使用氯硅烷;另一方面,本發明通過工藝控制與輔助催化劑的作用提高了產品的收率和選擇性;同時,本發明還具有成本低廉、操作簡捷、效率高、安全性好等優點。術語解釋:d4:八甲基環四硅氧烷的簡稱。d4vi:四乙烯基四甲基環四硅氧烷的簡稱。gc-ms:氣相色譜-質譜聯用技術。本發明的技術方案如下:一種2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷的制備方法,包括步驟:(1)在共裂解催化劑和輔助催化劑存在下,使八甲基環四硅氧烷(d4)和四乙烯基四甲基環四硅氧烷(d4vi)于常壓、90-110℃和攪拌條件下進行預裂解反應,反應時間1-2h;所述八甲基環四硅氧烷(d4)和四乙烯基四甲基環四硅氧烷(d4vi)按照摩爾比2:1;所述的共裂解催化劑為氫氧化鉀,輔助催化劑為三苯基膦;(2)向步驟(1)反應體系中補加共裂解催化劑,并升溫至140-150℃,進行共裂解反應,反應時間為0.5-1h;然后,(3)保持溫度140-150℃,將反應體系由常壓狀態轉變為負壓狀態,繼續進行共裂解反應,同時將反應生成的粗品采出,即收集70-80℃的餾分;在粗品采出的同時,將八甲基環四硅氧烷(d4)和四乙烯基四甲基環四硅氧烷(d4vi)按照摩爾比2:1同步滴加進反應體系,保持反應體系中的d4與d4vi的摩爾比始終為2:1,d4和d4vi的滴加總量與粗品采出總量保持在1:1質量比,滴加過程維持共裂解的溫度與壓力不變;(4)將步驟(3)收集的70-80℃餾分在真空條件下進行精餾,得到2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷。根據本發明,優選的,步驟(1)中的共裂解催化劑投料量為d4和d4vi投料總質量的0.5%-1%。步驟(1)中輔助催化劑投料量為d4和d4vi投料總質量的5%-10%。根據本發明,優選的,步驟(1)所述預裂解反應溫度為90-95℃。根據本發明,步驟(2)中所述補加共裂解催化劑的投料量為d4和d4vi投料總質量的0.5%-1%。根據本發明,優選的,步驟(2)中,升溫至145℃,共裂解反應時間為0.5h。根據本發明,優選的,步驟(3)中是采用水噴射真空泵將反應體系由常壓狀態改變為負壓狀態。根據本發明,優選的,步驟(3)中所述負壓狀態的壓力為-0.090mpa至-0.095mpa;進一步優選的,負壓狀態的壓力為-0.092mpa;粗品采出收集70-80℃/-0.092mpa的餾分。根據本發明,優選的,步驟(3)中d4與d4vi滴加過程的共裂解溫度保持140-150℃,共裂解壓力保持-0.090mpa至-0.095mpa。根據本發明,步驟(4)所述真空條件是壓力為-0.088mpa至-0.092mpa;所述精餾的回流比優選為7~8:1。本發明提供一種優選的實施方式,包括步驟如下:(1)在反應釜中加入592g八甲基環四硅氧烷和344g四乙烯基四甲基環四硅氧烷,混合均勻,然后向其中加入4.68g固體氫氧化鉀、56.16g三苯基膦,攪拌形成懸浮液;開啟加熱,緩慢升溫至95-100℃,進行充分的預裂解反應,反應時間1.5h;然后,(2)向釜內再加入4.68g固體氫氧化鉀,升溫至145℃,進行共裂解反應,反應時間0.5h;(3)采用水噴射真空泵,將體系由常壓狀態改變為負壓狀態,壓力維持-0.092mpa,繼續進行共裂解反應,收集70-80℃/-0.092mpa的餾分;在采出餾分的同時,向釜內按照摩爾比2:1同步滴加八甲基環四硅氧烷(d4)和四乙烯基四甲基環四硅氧烷(d4vi),保持體系中的d4與d4vi的摩爾比始終處于2:1,保持滴加的d4與d4vi的總量與采出的餾分總量為1:1質量比,整個過程保持釜溫145℃、壓力-0.092mpa不變;(4)將步驟(3)采出的餾分收集,在-0.090mpa的壓力下,采用8:1的回流比進行精餾提純,得2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷。產品純度大于99.5%,總收率大于83%。本發明沒有詳盡說明的,均按本領域常規技術。與現有技術相比,本發明的技術特點及有益效果如下:1、本發明是一種新的簡便的2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷的環保制備方法,該方法直接利用d4和d4vi裂解制備2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷;步驟(1)中d4和d4vi初期投料摩爾比為2:1,步驟(3)中d4和d4vi的同步滴加摩爾比為2:1,共裂解反應時70-80℃的餾分中二甲基硅氧鏈節摩爾總數與乙烯基甲基硅氧鏈節摩爾總數為2:1,通過同步滴加與采出的工藝控制,保證體系中的d4與d4vi的摩爾比基本恒定,始終處于2:1,在輔助催化劑三苯基膦的作用下,該連續過程抑制了環四硅氧烷的生成,同時極大的提高了環三硅氧烷的成環選擇性。2、本發明的方法制備的2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷收率高,原料易得,產品純度高,廢水量少。成本低廉。3、本發明不使用氯硅烷原料,解決了現有技術中使用氯硅烷帶來的環境污染問題;同時,本發明也不使用溶劑乙醚和三乙胺催化劑,避免反應過程產生大量的副產物三乙胺鹽酸鹽。4、本發明的方法共裂解過程中抑制了環四硅氧烷的生成,在提高了環三硅氧烷的成環選擇性的同時也避免了反應原料的爆聚、交聯,具有操作簡捷、效率高、安全性好等優點。附圖說明圖1是本發明實施例1共裂解采出的餾分的氣相色譜圖。圖2是本發明實施例1制備的產品2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷的1h-nmr譜圖。具體實施方式下面結合以下實施例對本發明作進一步說明,并不能限制本發明的內容。實施例中所述的原料均為常規原料,可市場購得或按照現有技術制備得到。實施例1、一種2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷的制備方法,包括步驟:(1)在裝有機械攪拌、恒壓滴液漏斗、溫度計和蒸餾塔的2l四口燒瓶中,加入592g(2mol)八甲基環四硅氧烷(d4)和344g(1mol)四乙烯基四甲基環四硅氧烷(d4vi),混合均勻,然后向其中加入4.68g固體氫氧化鉀、56.16g三苯基膦,攪拌形成懸浮液;開啟加熱,緩慢升溫,釜內物料逐漸變為粘稠狀,并伴有白色固體懸浮物,升溫至95-100℃時,體系逐漸由黏稠變稀,進行充分的預裂解反應,反應時間1.5h;然后,(2)向釜內再加入4.68g固體氫氧化鉀,升溫至145℃,進行共裂解反應,反應時間0.5h;(3)采用水噴射真空泵,將體系由常壓狀態改變為負壓狀態,壓力維持-0.092mpa,繼續進行共裂解反應,此時共裂解產生的餾分由蒸餾塔塔頂采出,收集70-80℃/-0.092mpa的餾分;在采出餾分的同時,由兩支恒壓滴液漏斗向釜內按照摩爾比2:1同步滴加八甲基環四硅氧烷(d4)和四乙烯基四甲基環四硅氧烷(d4vi),保持體系中的d4與d4vi的摩爾比始終處于2:1,并根據滴加與采出速率,保持滴加的d4與d4vi的總質量與采出的餾分質量為1:1,整個過程保持釜溫145℃、壓力-0.092mpa不變;(4)將步驟(3)采出的餾分收集,在-0.090mpa的壓力下,采用8:1的回流比進行精餾提純,得到產品2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷,純度99.55%,總收率83.4%。從步驟(3)共裂解采出的餾分取樣,使用gc-ms分析,氣相色譜圖如圖1所示,各組分的含量如下表1所示:表1組分名稱組分含量/%六甲基環三硅氧烷5.942-乙烯基-2,4,4,6,6-五甲基環三硅氧烷84.662,4-二乙烯基-2,4,6,6-四甲基環三硅氧烷8.18八甲基環四硅氧烷1.22步驟(4)產品的1h-nmr譜圖,如圖2所示,在氫譜的最高場0.2ppm處的峰組,積分面積為15,對應五個甲基,應為2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷中五個si-ch3,5.9ppm處為典型的乙烯基峰型,積分面積分別為1和2,對應si-ch=ch2中的三個氫原子。實施例2、一種2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷的制備方法,包括步驟:(1)在裝有機械攪拌、恒壓滴液漏斗、溫度計和蒸餾塔的2l反應釜中,加入592g(2mol)八甲基環四硅氧烷(d4)和344g(1mol)四乙烯基四甲基環四硅氧烷(d4vi),混合均勻,然后向其中加入4.68g固體氫氧化鉀、74.88g三苯基膦,攪拌形成懸浮液;開啟加熱,緩慢升溫,釜內物料逐漸變為粘稠狀,并伴有白色固體懸浮物,升溫至100℃時,體系逐漸由黏稠變稀,維持1-2h,進行充分的預裂解反應;然后,(2)向釜內再加入5.62g固體氫氧化鉀,升溫至143-147℃,維持40min,進行共裂解反應;(3)采用水噴射真空泵,將體系由常壓狀態改變為負壓狀態,壓力維持-0.092mpa繼續進行共裂解反應,此時共裂解產生的餾分由蒸餾塔塔頂采出,收集70-80℃/-0.092mpa的餾分;在采出餾分的同時,由兩支恒壓滴液漏斗向釜內按照摩爾比2:1同步滴加八甲基環四硅氧烷(d4)和四乙烯基四甲基環四硅氧烷(d4vi),保持體系中的d4與d4vi的摩爾比始終處于2:1,并根據滴加與采出速率,保持滴加的d4與d4vi的總質量與采出的餾分質量為1:1,整個過程保持釜溫143-147℃、壓力-0.092mpa不變;(4)將步驟(3)采出的餾分匯總,在-0.090mpa的壓力下,采用8:1的回流比進行精餾提純,得到產品2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷,純度99.74%,總收率81.3%。從步驟(3)共裂解采出的餾分取樣,使用gc-ms分析各組分的含量,結果如下表2所示:表2組分名稱組分含量/%六甲基環三硅氧烷7.232-乙烯基-2,4,4,6,6-五甲基環三硅氧烷83.782,4-二乙烯基-2,4,6,6-四甲基環三硅氧烷8.03八甲基環四硅氧烷0.96實施例3、一種2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷的制備方法,包括步驟:(1)在裝有機械攪拌、恒壓滴液漏斗、溫度計和蒸餾塔的2l反應釜中,加入592g(2mol)八甲基環四硅氧烷(d4)和344g(1mol)四乙烯基四甲基環四硅氧烷(d4vi),混合均勻,然后向其中加入5.62g固體氫氧化鉀、84.24g三苯基膦,攪拌形成懸浮液;開啟加熱,緩慢升溫,釜內物料逐漸變為粘稠狀,并伴有白色固體懸浮物,升溫至90-110℃時,體系逐漸由黏稠變稀,維持1-2h,進行充分的預裂解反應;然后,(2)向釜內再加入5.62g固體氫氧化鉀,升溫至140-150℃,維持0.5h,進行共裂解反應;(3)采用水噴射真空泵,將體系由常壓狀態改變為負壓狀態,壓力維持-0.092mpa繼續進行步驟(2)共裂解反應,此時共裂解產生的餾分由蒸餾塔塔頂采出,收集70-80℃/-0.092mpa的餾分;在采出餾分的同時,由兩支恒壓滴液漏斗向釜內按照摩爾比2:1同步滴加八甲基環四硅氧烷(d4)和四乙烯基四甲基環四硅氧烷(d4vi),保持體系中的d4與d4vi的摩爾比始終處于2:1,并根據滴加與采出速率,保持滴加的d4與d4vi的總質量與采出的餾分質量為1:1,整個過程保持釜溫140-150℃、壓力-0.092mpa不變;(4)將步驟(3)采出的餾分匯總,在-0.090mpa的壓力下,采用8:1的回流比進行精餾提純,得到產品2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷,純度99.53%,總收率82.9%。從步驟(3)共裂解采出的餾分取樣,使用gc-ms分析各組分的含量,結果如下表3所示:表3組分名稱組分含量/%六甲基環三硅氧烷7.052-乙烯基-2,4,4,6,6-五甲基環三硅氧烷84.012,4-二乙烯基-2,4,6,6-四甲基環三硅氧烷7.85八甲基環四硅氧烷1.09比較例1(1)在裝有機械攪拌、溫度計和蒸餾塔的2l反應釜中,加入592g(2mol)八甲基環四硅氧烷(d4)和344g(1mol)四乙烯基四甲基環四硅氧烷(d4vi),混合均勻,然后向其中加入4.68g固體氫氧化鉀,攪拌形成懸浮液;開啟加熱,緩慢升溫,釜內物料逐漸變為粘稠狀,并伴有白色固體懸浮物,升溫至90-110℃時,體系十分粘稠,維持1-2h;(2)向釜內再加入4.68g固體氫氧化鉀,升溫至140-150℃,體系仍舊呈現明顯的粘滯狀態;(3)采用水噴射真空泵,將體系抽至負壓,壓力維持-0.090mpa至-0.095mpa繼續進行反應,塔頂僅有少量的餾分出現;(4)采集餾分的過程中,釜內物料變為暗褐色,粘度進一步增加,并有交聯現象發生,隨著反應進行,體系交聯明顯,釜內物料結塊,攪拌困難,塔頂幾乎無餾分。裂解采出的餾分,使用gc-ms分析各組分的含量,結果如下表4所示:表4組分名稱組分含量/%六甲基環三硅氧烷21.852-乙烯基-2,4,4,6,6-五甲基環三硅氧烷10.622,4-二乙烯基-2,4,6,6-四甲基環三硅氧烷7.91八甲基環四硅氧烷30.762,4,6-三乙烯基-2,4,6-三甲基環三硅氧烷8.662-乙烯基-2,4,4,6,6,8,8-七甲基環四硅氧烷11.212,4-二乙烯基-2,4,6,6,8,8-六甲基環四硅氧烷5.672,4,6,8-四乙烯基-2,4,6,8-四甲基環四硅氧烷1.46比較例1雖然是按照八甲基環四硅氧烷(d4)和四乙烯基四甲基環四硅氧烷(d4vi)摩爾比2:1投料,但是未加輔助催化劑三苯基膦。在共裂解的過程中,釜內物料十分黏稠,結膠明顯,僅僅裂解出極少量的餾分,收率很低;并且餾分的組分(如表4)十分復雜,2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷的選擇性很低,無法高效的合成產品。比較例2(1)在裝有機械攪拌、溫度計和蒸餾塔的2l四口燒瓶中,加入296g(1mol)八甲基環四硅氧烷(d4)和344g(1mol)四乙烯基四甲基環四硅氧烷(d4vi),混合均勻,然后向其中加入3.4g固體氫氧化鉀、470.4g二苯醚和169.6g聯苯,攪拌形成懸浮液;(2)開啟加熱,緩慢升溫,同時對體系進行抽真空處理,壓力維持在-0.097mpa至-0.099mpa,升溫至130℃,進行裂解反應;(3)繼續步驟(2)的裂解反應,采集塔頂80-100℃的餾分。裂解采出的餾分,使用gc-ms分析各組分的含量,結果如下表5所示:表5組分名稱組分含量/%六甲基環三硅氧烷1.852-乙烯基-2,4,4,6,6-五甲基環三硅氧烷3.66八甲基環四硅氧烷10.762-乙烯基-2,4,4,6,6,8,8-七甲基環四硅氧烷28.252,4-二乙烯基-2,4,6,6,8,8-六甲基環四硅氧烷53.86比較例2是按照八甲基環四硅氧烷(d4)和四乙烯基四甲基環四硅氧烷(d4vi)摩爾比1:1投料,選用二苯醚和聯苯的混合物作為導熱介質,進行共裂解反應。在更改投料比例及缺少本發明輔助催化劑的情況下,如表5所示,共裂解產生的餾分主要為環四硅氧烷,2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷的含量很低,選擇性很差。比較例3(1)在裝有機械攪拌、溫度計和蒸餾塔的2l四口燒瓶中,加入758g(1mol)二甲基硅油(ho-[si(ch3)2o]10-h)和878g(1mol)甲基乙烯基硅油(ho-[si(ch3)(ch=ch2)o]10-h),混合均勻,然后向其中加入16.36g固體氫氧化鉀、164g礦物油,攪拌形成懸浮液;(2)開啟加熱,緩慢升溫,同時對體系進行抽真空處理,壓力維持在-0.097mpa至-0.099mpa,升溫至150-160℃,進行裂解反應;(3)收集步驟(2)裂解反應產生的餾分。裂解采出的餾分,使用gc-ms分析各組分的含量,結果如下表6所示:表6組分名稱組分含量/%六甲基環三硅氧烷3.062-乙烯基-2,4,4,6,6-五甲基環三硅氧烷1.22八甲基環四硅氧烷17.632-乙烯基-2,4,4,6,6,8,8-七甲基環四硅氧烷26.342,4-二乙烯基-2,4,6,6,8,8-六甲基環四硅氧烷35.782,4,6-三乙烯基-2,4,6,8,8-五甲基環四硅氧烷13.44比較例3是采用二甲基硅油和甲基乙烯基硅油作為裂解原料,其中二甲基硅氧鏈節和甲基乙烯基硅氧鏈節的摩爾比為1:1,取代本發明的輔助催化劑,改用礦物油,進行共裂解反應。餾分結果如表6所示,裂解產物主要為各種環四硅氧烷,2-乙烯基-2,4,4,6,6-五甲基環三硅氧烷的收率與選擇性均很低,無法有效得到。比較例4(1)向2l四口燒瓶中加入1000g20%的鹽酸,劇烈攪拌,然后由兩支恒壓滴液漏斗同步滴加258g(2mol)二甲基二氯硅烷和141g(1mol)甲基乙烯基二氯硅烷,進行共水解反應。共水解的溫度維持10-50℃,滴加完成后,繼續攪拌2h,然后靜置、分層,將有機相水洗至中性,干燥后得到共水解物;(2)在裝有機械攪拌、溫度計和蒸餾塔的另一2l四口燒瓶中,加入180g上述的共水解物、1.8g固體氫氧化鉀、20g十八醇,攪拌均勻;(3)開啟加熱,緩慢升溫,同時對體系進行抽真空處理,壓力維持在-0.090mpa至-0.095mpa;(4)升溫至55℃時,體系爆聚,釜溫即由55℃迅速沖溫至102℃,釜內物料交聯明顯,由油狀物變為固體膠渣,無法裂解,無餾分采出。比較例4是按照二甲基二氯硅烷和甲基乙烯基二氯硅烷摩爾比2:1進行共水解反應,旨在合成摩爾比2:1的共水解物,然后將共水解物進行裂解處理。反應過程中,體系爆聚,物料交聯劇烈。該情況的出現,說明共水解物中組分成分復雜,含有各種環體及直鏈聚硅氧烷,在強堿的作用下,環體開環聚合,反應大量放熱,并引起乙烯基的交聯發生,熱量進一步疊加,最終致使整個體系爆聚,無法裂解。當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1