燈盞乙素生物素標記探針的應用及相關PKM2激酶抑制劑的制作方法
本發明涉及醫藥技術領域,具體涉及燈盞乙素苷元生物素標記探針的制備、用途和可用作pkm2激酶抑制劑的燈盞乙素苷元及其衍生物,包含此化合物的藥物組合物,涉及所述化合物的制備方法及所述化合物在制備m2型丙酮酸激酶抑制劑中的應用,涉及所述化合物在疾病,例如pkm2過表達的癌癥治療中的用途。
背景技術:
靶向治療是利用腫瘤細胞可以表達,而正常細胞很少或不表達的特定基因或基因的表達產物,形成相對或絕對靶向,最大限度地殺傷腫瘤細胞,而對正常細胞損傷很小的治療方法。目前越來越多的研究者把注意力轉向了來源豐富、價廉易得、毒副作用小、分子結構多樣的天然產物,并且對天然產物維護人類健康或預防疾病充滿了期望。相繼也發現了一些具有藥理活性的有效成分,如發現黃酮類、萜類、生物堿類具有抗腫瘤的作用。近年來從藥用植物提取物的黃酮類化合物的抗癌活性已報道(liweberm.newtherapeuticaspectsofflavones:theanticancerpropertiesofscutellariaanditsmainactiveconstituentswogonin,baicaleinandbaicalin[j].cancertreatmentreviews,2009,35(1):57.),燈盞乙素(scutellarin)是從菊科植物燈盞細辛中提取的黃酮類化合物。人體藥動學藥效學研究(gaoc,zhangh,guoz,etal.mechanisticstudiesontheabsorptionanddispositionofscutellarininhumans:selectiveoatp2b1-mediatedhepaticuptakeisalikelykeydeterminantforitsuniquepharmacokineticcharacteristics[j].drugmetabolism&disposition,2012,40(10):2009.)表明了燈盞乙素7位的葡萄糖在體內代謝發生水解成燈盞乙素苷元(scutellarein,sc)而被吸收,并且燈盞乙素苷元的藥效同燈盞乙素相似,燈盞乙素苷元是燈盞乙素發揮藥理作用的主要形式。
近期研究表明燈盞乙素苷元具有廣泛的抗腫瘤活性(fengy,zhangs,tuj,etal.novelfunctionofscutellarinininhibitingcellproliferationandinducingcellapoptosisofhumanburkittlymphomanamalwacells[j].leukemia&lymphoma,2012,53(12):2456.),然而,其直接作用靶點蛋白和抗腫瘤作用機制尚不明確。因此設計合成燈盞乙素生物素苷元標記探針,以探針為工具,運用蛋白質組學,以期發現燈盞乙素苷元發揮抗腫瘤活性的直接靶點蛋白,一旦可能的靶標被發現后,再通過一系列的分子生物學實驗來確證這些靶點蛋白及其生物學作用。
本發明以探針為工具和手段,利用蛋白質組學發現燈盞乙素苷元抗腫瘤作用的靶點為pkm2。m2型丙酮酸激酶(pkm2)是糖酵解中的一個關鍵限速酶,在能量代謝中起著關鍵的作用(mazureks.pyruvatekinasetypem2:akeyregulatorofthemetabolicbudgetsystemintumorcells[j].internationaljournalofbiochemistry&cellbiology,2011,43(7):969.)。因此,以pkm2為靶點的藥物代表針對特定分子靶點的新一代靶向治療,且因此與常規化學治療相比能在治療各種癌癥方面提供更大功效且同時具有更小副作用,為后續靶向藥物開發提供重要理論依據,從而開發出藥效更好,特異性更強的靶向藥物。
技術實現要素:
本發明通過設計合成燈盞乙素苷元生物素標記探針并考察探針和燈盞乙素苷元抗腫瘤活性,以探針為工具,運用蛋白質組學,發現燈盞乙素苷元發揮抗腫瘤活性的直接靶點蛋白,一旦可能的靶標被發現后,再通過一系列的分子生物學實驗來確證這些靶點蛋白及其生物學作用。
本發明利用燈盞乙素苷元生物素標記探針尋找到燈盞乙素苷元用于治療癌癥的靶點pkm2蛋白,重組了pkm2蛋白,檢測了化合物對重組pkm2活性影響,針對靶點pkm2蛋白對燈盞乙素苷元結構改造來尋找新型高效低毒的選擇性pkm2調節劑。
因此,本發明的目的是提供用燈盞乙素苷元為先導化合物與wr試劑和勞森試劑反應成燈盞乙素苷元衍生物的制備方法和應用,可以解決現有技術存在的抑制pkm2活性不高的問題,而且本發明制得的燈盞乙素苷元衍生物水溶性、對人成骨肉瘤saos-2細胞等多種腫瘤細胞生長抑制均較好,可應用于制備預防或治療腫瘤的藥物。
本發明是這樣實現的:
本發明提出了具有通式(ⅰ)的燈盞乙素苷元生物素標記探針的結構模塊:
該類結構模塊包括活性基團、鏈接基團、報告集團,r可以是長烷烴鏈、聚乙二醇鏈、多肽鏈。本發明主要以常見的2,2'-(乙烯二氧)雙(乙胺)為例,其中r為無時,得到探針p4;r為2,2'-(乙烯二氧)雙(乙胺),得到探針p10。
因此,由該結構模塊制備得到的燈盞乙素苷元生物素標記探針結構是下列任一化合物:
該燈盞乙素苷元生物素標記探針的制備方法是:燈盞乙素苷元5、6、7、4'的羥基上的氫原子被二苯羧酮保護后,燈盞乙素苷元直接或間接與生物素相連,最后在tfa的作用下脫去保護基團,得到兩個不同的燈盞乙素生物素標記探針。
該燈盞乙素苷元生物素標記探針的用途時:該探針用于尋找燈盞乙素苷元治療癌癥的靶點pkm2蛋白,從而合成作為pkm2激酶抑制劑的燈盞乙素苷元衍生物及其藥學上可接受的鹽。
該燈盞乙素苷元衍生物及其藥學上可接受的鹽,具有通式(ⅱ)所示的結構:
其中r1為me時,x為o;r1為h時,x為o;r1為me時,x為s;r1為h時,x為s;r1為me時,x為se;r1為h時,x為se。
具體的,該燈盞乙素苷元衍生物及其藥學上可接受的鹽,其結構選自:
該燈盞乙素苷元衍生物及其藥學上可接受的鹽的制備方法是:燈盞乙素苷元5、6、7、4'的羥基上的氫原子被甲基取代后,3位上的氧分別被勞森試劑和wr試劑取代為含硫和硒的中間體,最后在bbr3的作用下脫去保護基團甲基,得到兩個與燈盞乙素苷元不同的衍生物。
本發明還提出了該燈盞乙素苷元衍生物及其藥學上可接受的鹽在制備治療腫瘤的藥物中的應用。其中,所述藥物為pkm2激酶抑制劑類抗腫瘤藥物。
與現有技術相比,本發明突出的實質性特點和顯著的進步在于:本發明提供了一種m2型丙酮酸激酶(pkm2)抑制劑燈盞乙素苷元生物素標記探針及其衍生物的制法和探針在靶點蛋白尋找的應用、驗證,該衍生物是從燈盞細辛中提取分離得到的燈盞乙素苷元為先導化合物與wr試劑和勞森試劑反應制得的。成藥性初步研究表明本發明的燈盞乙素苷元生物素標記探針及其衍生物對pkm2抑制強度與紫草素類似,比燈盞乙素苷元提高約20或40多倍,能選擇性抑制腫瘤細胞增殖,而對正常細胞(hek293)幾乎沒有影響,屬于新型pkm2抑制劑類抗腫瘤藥物,尤其是當o被se取代后,所以藥理作用最為明顯。本發明原料來源豐富,工藝簡捷,純度高,成本低,高效低毒,有望成為新型高效低毒的pkm2抑制劑類抗癌藥物。
附圖說明
圖1是本發明的探針p4制備路線;
圖2是本發明的探針p10制備路線;
圖3是本發明的燈盞乙素苷元衍生物的制備路線;
圖4是本發明的通過質譜鑒定pkm2為燈盞乙素的潛在靶點蛋白(a);
圖5是本發明的通過質譜鑒定pkm2為燈盞乙素的潛在靶點蛋白(b);
圖6是本發明的探針10與pkm2蛋白相互作用;
圖7是本發明的丙酮酸激酶活性測定的示意圖。
具體實施方式
本發明的燈盞乙素苷元生物素標記探針的結構模塊具有以下結構式:
式(ⅰ)中母核結構為燈盞乙素苷元,其中無r=0時,為探針p4,r=2,2'-(乙烯二氧)雙(乙胺)時,為探針p10。
本發明的燈盞乙素苷元衍生物及其藥學上可接受的鹽具有以下結構式:
式(ⅱ)中母核結構為5燈盞乙素苷元化合物,其中,r1為me,x為o、s、se的雜原子;r1為h,x為o、s、se的雜原子。
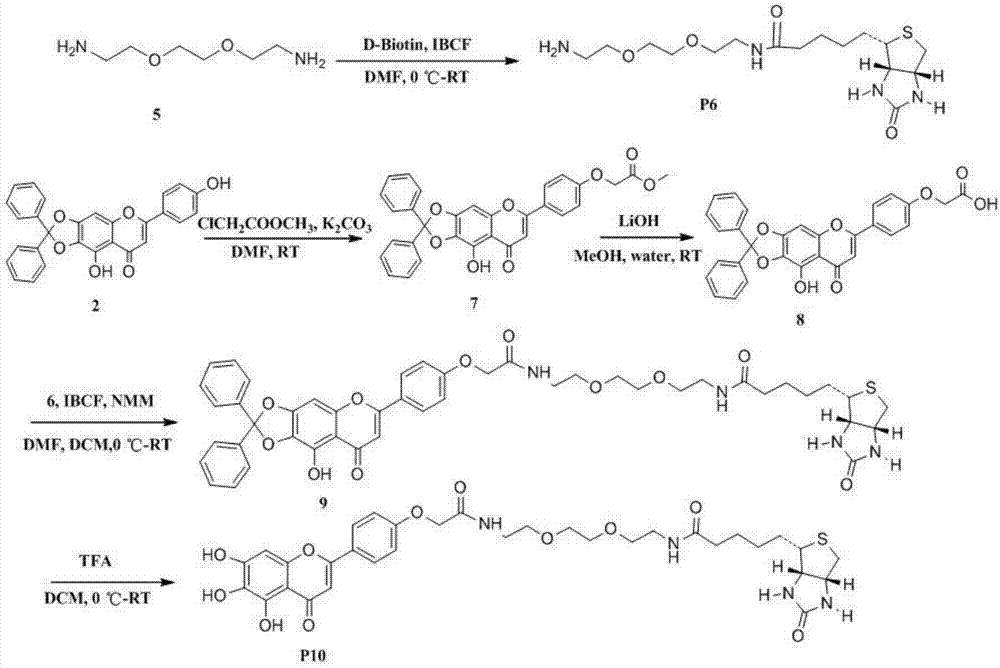
燈盞乙素生物素標記探針的制備原理:燈盞乙素苷元5、6、7、4'的羥基上的氫原子被二苯羧酮保護后,燈盞乙素苷元直接或間接與生物素相連,最后在tfa的作用下脫去保護基團,得到兩個不同的燈盞乙素生物素標記探針。如圖1和圖2所示。
燈盞乙素苷元衍生物的制備原理:燈盞乙素苷元5、6、7、4'的羥基上的氫原子被甲基取代后,3位上的氧分別被勞森試劑和wr試劑取代為含硫和硒的中間體,最后在bbr3的作用下脫去保護基團甲基,得到兩個與燈盞乙素苷元不同的衍生物。如圖3所示。
以下結合實施例對本發明作進一步的詳細描述,本發明的保護范圍并不僅限于此:
實施例1:
縮酮保護燈盞乙素苷元(2)的制備:
取干燥的燈盞乙素苷元(1)(0.5g,1.75mmol)置于干燥的100ml二頸瓶中,加入10mldme溶解,加入dmap(0.25g,1.75mmol),室溫攪拌下加入二氯二苯甲烷(0.5ml,1.75mmol),180℃條件下,回流反應2h,tlc監測反應完全,減壓旋蒸除去溶劑,硅膠分離殘余物,得淺棕黃色粉末狀固體(2),0.34g,收率為62%。1h-nmr(400mhz,dmso-d6)δ(ppm):7.92(d,j=8.8hz,2h),7.56-7.53(m,4h),7.46-7.44(m,6h),7.04(s,1h),6.90(d,j=8.8hz,2h),6.83(s,1h).ei-ms(m/z)[m+h]+451.11。
燈盞乙素苷元-4'-生物素(3)的制備:
取縮酮保護燈盞乙素苷元(2)(0.4g,0.89mmol)于100ml的茄型瓶中,分別依次加入dmap(0.054g,0.44mmol),生物素(0.433g,1.78mmol),室溫攪拌至完全溶解后,再緩慢滴加用dmf溶解的dcc(0.366g,1.78mmol),滴加完畢,室溫反應12h,tlc監測反應完全,用乙酸乙酯(150ml×2)萃取,有機層用水(100ml×2)洗滌,無水mgso4干燥,減壓回收乙酸乙酯至干,硅膠柱層析得到淡黃色粉末狀固體(3)0.2g,收率為30%。1h-nmr(400mhz,dmso-d6)δ(ppm):8.13(d,j=8.0hz,2h),7.56-7.43(m,10h),7.34(d,j=8.0hz,2h),7.12(s,1h),7.06(s,1h),6.47(s,1h),6.38(s,1h),2-4.29m,1h),4.16-4.13(m,1h),3.50-3.44(m,1h),3.42-3.37(m,1h),3.15-3.11(m,1h),2.87-2.81(m,1h),2.71-2.49(m,3h),1.67-1.63(m,3h),1.56-1.37(m,2h).ei-ms(m/z)[m+h]+677.73。
脫縮酮保護燈盞乙素苷元-4'-生物素(4)的制備:
取化合物(3)10mg于50ml的茄型瓶中,加入5ml干燥的ch2cl2,冰浴條件下加入2mltfa,加畢,室溫反應17h,減壓蒸除溶劑,用乙酸乙酯沉降3次,得到黃色固體(4)8mg.收率為85%。1h-nmr(400mhz,dmso-d6)δ(ppm):8.12(d,j=12hz,2h),7.32(d,j=12hz,2h),6.93(s,1h),6.61(s,1h),6.48(s,1h),6.38(s,1h),4.32-4.29(m,1h),4.16-4.13(m,1h),3.17-3.11(m,2h),2.85-2.81(m,1h),2.64-2.48(m,2h),1.69-1.64(m,2h),1.53-1.40(m,2h).13cnmr(dmso-d6,400mhz):182.11,171.53,162.73,162.22,153.64,153.10,149.80,146.95,128.51,127.89,122.65,104.49,104.22,94.03,61.02,59.20,55.34,48.61,33.32,28.00,27.93,24.30.ei-ms(m/z)[m+h]+513.13.hrmscalcdforc25h23n2o8s-[m-h]-511.1223,found:511.1228。
長鏈生物素(6)的制備:
取生物素(600mg,2.5mmol)和三乙胺(0.38ml,3mmol)置于100ml茄型瓶中,用無水dmf將其溶解,然后在冰浴下攪拌半個小時,然后緩慢滴加氯甲酸異丁酯(0.44ml,3mmol)加畢,體系在冰浴下反應3小時,然后將反應液緩慢滴加到2,2'-(乙烯二氧)雙(乙胺)(655.14mg,4.4mmol)的dmf溶液中,加畢,室溫反應4個小時。反應完后用旋蒸儀蒸出dmf,過柱得到產物(6)10mg,收率50%。1h-nmr(400mhz,dmso-d6)δ(ppm):8.909(m,1h),6.35(m,2h),4.27-4.24(m,1h),4.10-4.09(m,1h),3.58-3.33(m,9h),3.24-3.03(m,5h),2.92-2.89(m,2h),2.17-2.14(m,2h),1.56-1.26(m,7h).ei-ms(m/z)[m+h]+375.50。
縮酮保護燈盞乙素苷元-4'-氯乙酸甲酯(7)的制備:
取縮酮保護燈盞乙素苷元(2)(0.50g,1.11mmol)與無水k2co3(0.31g,2.22mmol)和clch2cooch3(0.14g,1.33mmol)混合于反應瓶中,氮氣保護下注入無水dmf15ml,室溫攪拌反應過夜。經萃取、洗滌、干燥后,減壓回收溶劑至干得(7)0.37g,收率為64%。1h-nmr(400mhz,dmso-d6)δ(ppm):8.03(d,j=8hz,2h),7.55-7.46(m,10h)7.12(d,j=8hz,2h),6.98(s,1h),6.72(s,1h),5.75(s,1h),4.98-4.92(m,2h),3.70(s,3h).ei-ms(m/z)[m+h]+523.50。
縮酮保護燈盞乙素苷元-4'-氯乙酸甲酯水解(8)的制備:
取上述所得的化合物(7)溶解于甲醇和水(甲醇:水=9:1),室溫下緩慢加入用甲醇溶解的氫氧化鋰(20.35mg,0.8mmol),室溫反應3h,反應完畢,經萃取、洗滌、干燥后,減壓回收溶劑至干得(8)0.30g,收率為90%。1h-nmr(400mhz,dmso-d6)δ(ppm):8.02(d,j=8hz,2h),7.56-7.40(m,10h),7.03(d,j=8hz,2h),6.98(s,1h),6.72(s,1h),4.59(s,2h).ei-ms(m/z)[m+h]+509.48。
縮酮保護燈盞乙素苷元-4'-長鏈生物素(9)的制備:
取縮酮保護燈盞乙素苷元-4'-氯乙酸甲酯水解產物(8)0.30g和nmm(137.26mg,1.36mmol)于干燥的反應瓶中,用10ml無水dmf溶解,并在冰浴下反應半個小時,再緩慢加入含有ibcf(96.7mg,0.7mmol)的二氯甲烷,繼續在冰浴下反應3h,然后緩慢加入上述產物長鏈生物素(265.14mg,0.59mmol),加畢,體系在室溫反應20h,反應完畢,經萃取、洗滌、干燥后,減壓回收溶劑至干得黃色固體(9)100mg,收率20%。1h-nmr(400mhz,cd3cl)δ(ppm):7.80(d,j=8hz,2h),7.60-7.54(m,3h),7.38-7.35(m,7h),7.02(d,j=8hz,2h),7.15(s,1h),6.62-6.53(m,3h),6.46(s,1h),5.63(s,1h),4.56(s,2h),4.44(s,1h),4.25(s,1h),3.55(m,11h),3.38(m,2h),2.84(m,1h),2.79(m,1h),2,79-2,16(m,2h),1.65-1,54(m,5h)。13cnmr(cd3cl,400mhz):183.02,173.35,167.82,163.91,163.52,160.09,153.51,153.25,142.32,139.30,130.07,129.84,129.56,128.54,128.45,128.29,128.12,128.01,126.46,126.38,124.92,119.29,115.37,107.71,104.54,89.57,70.23,70.16,69.95,69.71,67.45,61.89,60.32,55.55,40.56,39.23,39.01,35.95,28.18,28.11,27.89,25.59.ei-ms(m/z)[m+h]+865.10.hrmscalcdforc46h47n4o11s-[m-h]-865.3155,found:865.3150。
脫縮酮保護燈盞乙素苷元-4′-長鏈生物素(10)的制備
10mg于10ml的茄型瓶中,加入0.5ml干燥的ch2cl2,冰浴條件下加入0.2ml三氟乙酸(tfa),加畢,室溫反應10h,減壓蒸除溶劑,用乙酸乙酯沉降3次,得到黃色固體(10)6mg,收率為60%。1h-nmr(400mhz,dmso-d)δ(ppm):8.04(d,j=6hz,2h),7.12(d,j=6hz,2h),6.86(s,1h),6.61(s,1h),4.62(m,2h),4.31-4.27(m,1h),4.12-4.09(m,1h),3.51-3.28(m,14h),3.20-3.17(m,1h),2.82-2.78(m,1h),2.07-2.03(m,2h),1.60-1.42(m,4h),1.32-1.23(m,2h).13cnmr(dmso-d,400mhz):181.93,172.01,167.14,162.57,153.32,146.90,128.03,115.16,104.09,102.95,93.82,69.41,69.06,68.71,67.45,60.92,59.07,55.31,54.80,40.5,39.21,39.00,34.97,28.08,27.92,25.24.ei-ms(m/z)[m+h]+701.60.hrmscalcdforc33h39n4o11s-[m-h]-699.2410,found:699.2416。
實施例2:
燈盞乙素苷元衍生物本發明自行制備,制備方法如下:
化合物sc2的制備:
取燈盞乙素苷元(1g,3.49mml)和k2co3(2.41g,17.47mml)在15ml丙酮溶液中,油浴至回流,緩慢滴加1.3ml,加畢,回流反應2h,tlc監測反應完全,用乙酸乙酯(300ml×2)萃取,有機層用水(300ml×2)洗滌,無水mgso4干燥,減壓回收乙酸乙酯至干,硅膠柱層析得到淡黃色粉末狀固體sc2(0.96g),收率為85%。1h-nmr(400mhz,dmso-d6)δ(ppm):7.92(d,j=8.8hz,2h),6.90(d,j=8.8hz,2h),6.83(s.1h),6.30(s.1h),3.95(s.12h).ei-ms(m/z)[m+h]+343.34。
化合物sc3的制備:
取化合物sc2(0.5g,1.46mml)和勞氏試劑(0.59g,1.46mml)在10mlthf溶液中,加畢,室溫反應3天,tlc監測反應完全,用乙酸乙酯(200ml×2)萃取,有機層用水(200ml×2)洗滌,無水mgso4干燥,減壓回收乙酸乙酯至干,硅膠柱層析得到粉末狀固體sc3(0.25g),收率為50%。1h-nmr(400mhz,dmso-d6)δ(ppm):7.92(d,j=8.8hz,2h),6.90(d,j=8.8hz,2h),5.63(s,1h),5.99(s,1h),3.95(s.12h).ei-ms(m/z)[m+h]+359.43。
化合物sc4的制備:
取化合物sc3(0.2g,0.558mml)在5mlch2cl2溶液中,在冰浴-20度下緩慢滴加bbr3(0.97ml,10.04mml),加畢,室溫反應6h,tlc監測反應完全,用乙酸乙酯(200ml×2)萃取,有機層用水(200ml×2)洗滌,無水mgso4干燥,減壓回收乙酸乙酯至干,硅膠柱層析得到粉末狀固體sc4(0.13g),收率為80%。1h-nmr(400mhz,dmso-d6)δ(ppm):7.86(d,j=8.8hz,2h),6.84(d,j=8.8hz,2h),5.63(s,1h),5.89(s,1h),5.53(s,4h).13cnmr(dmso-d6,400mhz):202.8,157.7,154.8,154.3,152.4,129.6,129.9,122.9,120.1,115.8,111.6,94.3.ei-ms(m/z)[m+h]+303.02.hrmscalcdforc25h23n2o8s-[m-h]-301.0223,found:301.0228。
化合物sc5的制備:
取化合物sc2(0.5g,1.46mml)和wr試劑(0.78g,1.46mml)在10ml乙腈溶液中,加畢,置于完全密封的微管中,在油浴中150℃下反應1h,tlc監測反應完全,用乙酸乙酯(200ml×2)萃取,有機層用水(200ml×2)洗滌,無水mgso4干燥,減壓回收乙酸乙酯至干,硅膠柱層析得到粉末狀固體sc5(0.4g),收率為83%。1h-nmr(400mhz,dmso-d6)δ(ppm):7.86(d,j=8.8hz,2h),6.84(d,j=8.8hz,2h),5.63(s,1h),5.89(s,1h),3.83(s,12h).ei-ms(m/z)[m+h]+407.03。
化合物sc6的制備:
取化合物sc5(0.2g,0.493mml)在5mlch2cl2溶液中,置于冰浴-20度下緩慢滴加bbr3(0.86ml,8.88mml),加畢,室溫反應10h,tlc監測反應完全,用乙酸乙酯(200ml×2)萃取,有機層用水(200ml×2)洗滌,無水mgso4干燥,減壓回收乙酸乙酯至干,硅膠柱層析得到粉末狀固體sc6(0.13g),收率為80%。1h-nmr(400mhz,dmso-d6)δ(ppm):7.86(d,j=8.8hz,2h),6.84(d,j=8.8hz,2h),5.63(s,1h),5.89(s,1h),5.53(s,4h).13cnmr(dmso-d6,400mhz):192.8,157.7,156.6,153.3,152.8,151.4,130.2,129.2,122.9,115.8,111.6,97.3,94.9,84.6.ei-ms(m/z)[m+h]+350.97.hrmscalcdforc25h23n2o8s-[m-h]-348.9223,found:348.9228。
探針p4、p10抗腫瘤活性:
探針p4、p10和sc1對人宮頸癌hela細胞在48h體外抗腫瘤活性測定結果。將上述三種待測化合物用dmso溶解成相應濃度,以培養液稀釋成終濃度分別為10μm-400μm,加入到相應的板孔中,每孔體積100μl,同時設置調零孔(加入等量培養基)、細胞對照組(加入不含藥物培養基)。置37℃,5%co2培養箱內培養48h后,每孔加入5μlmts,繼續培養4h后,將96孔板置于酶標儀中,在490nm波長處測定吸光度值(od),計算細胞存活率(結果見表1),每個濃度平行6孔,實驗重復3次,計算公式如下:
表1燈盞乙素苷元及探針體外抗腫瘤活性
以上結果說明,探針p4、p10與燈盞乙素苷元抗腫瘤活性相當,探針p4、p10能用來確定燈盞乙素苷元的抗腫瘤作用靶點蛋白。
探針p4和p10的用途:
先將化學探針p4和p10與蛋白質提取液進行孵育,生物素-鏈親和素層析分離技術的方法分離純化蛋白,通過高靈敏度的質譜鑒定這些候選蛋白,通過對質譜鑒定到的蛋白的細胞定位、功能和參與的生物學過程查詢,得到5個可能與探針p4和p10相互作用的靶點蛋白,詳細信息見表2,分別為55kd的pkm2、105kd的nfkb1、90kd的hsp90、36kd的gapdh、42kd的ndrg1。在所發現的靶點蛋白中,其中在55kda處有明顯的蛋白條帶(見圖4),根據進一步的質譜二級譜圖解析其唯一多肽片段表明該條帶可能為m2型丙酮酸激pkm2(見圖5)。
表2mascot分析lc–ms/ms鑒定p4和p10垂釣的蛋白
探針p10與pkm2蛋白相互作用驗證:
hela細胞經200μm探針p10處理6小時后,提取hela細胞裂解液,利用生物素與鏈霉素親和素之間高度的親和力,免疫共沉淀p10上面的生物素,蛋白印跡檢測pkm2蛋白,結果顯示(見圖6中a部分),hela、mcf-7、a549細胞均能ip出pkm2蛋白,證明p10和pkm2相互作用。生物素單體作為競爭性對照,通過競爭的方法抑制探針分子與靶點蛋白的結合,加入不同濃度的生物素可能會出現特征性條帶逐漸變淡的現象。進一步的免疫共沉淀結果顯示(見圖6中b部分),streptavidinbeads中pkm2隨生物素濃度增大條帶變淺,上清液中pkm2隨生物素濃度增大條帶變深,說明pkm2蛋白與p10相互作用為特異結合。
重組pkm1、pkm2方法如下:
top10(購自qiagen公司)中,經上海美吉生物公司測序,ncbi數據庫比對,所克隆出的基因確pkm2基因,通過iptg(購自qiagen公司)誘導表達出pkm2蛋白,經純化得到純度為99%的pkm2蛋白。pkm1、pkm2活性的測定:根據vanderheiden等[heidenmgv,christofkhr,schumane,etal.identificationofsmallmoleculeinhibitorsofpyruvatekinasem2[j].biochemicalpharmacology,
2010,79(8):1118.]人報道的方法進行酶的活性和抑制試驗。該法是由乳酸脫氫酶偶聯法測定丙酮酸酶的活性(見圖7),根據乳酸脫氫酶的底物nadh在340nm有吸收,可以通過紫外分光光度檢測nadh含量減少來判斷反應進行的速度。丙酮酸反應液成分是:trisph7.5(50mm),kcl(100mm),mgcl2(5mm),adp(0.6mm),pep(0.5mm),β-nadh(180mm),fbp(10mm)andldh(8units)。將上述制備的燈盞乙素苷元及其衍生物sc1,sc2,sc3,sc4,sc5,sc6配成濃度0.25mm,0.5mm,1mm,2mm,4mm,8mm,16mm,32mm,64mm的母液。酶活檢測的過程如下:先將20-100ng/μl的重組pkm2蛋白和相應濃度fbp預孵育30分鐘,然后將重組pkm2蛋白與相應濃度的藥物混合(反應體系中dmso濃度不能超過1%體積比,避免dmso影響酶活),室溫孵育30分鐘,再將800μl的孵育液與200μl酶反應液在比色皿中迅速混合,插入紫外分光光度計檢測槽,檢測od340nm在5-10分鐘的降低變化,程序設定每0.1秒記錄一次吸光值。根據一定時間內吸光值的變化,其吸光值和濃度呈線性正比關系,比較對照組和藥物處理組酶活性,od340nm變化越慢則表示酶活越低。
燈盞乙素苷元及其衍生物抗腫瘤活性篩選:
燈盞乙素苷元及其衍生物sc1、sc4、sc6對人成骨肉瘤saos-2細胞、人宮頸癌hela細胞等10種腫瘤細胞在48h體外抗腫瘤活性測定結果。將上述三種待測化合物用dmso溶解成相應濃度,以培養液稀釋成終濃度分別為10μm-400μm,加入到相應的板孔中,每孔體積100μl,同時設置調零孔(加入等量培養基)、細胞對照組(加入不含藥物培養基)。置37℃,5%co2培養箱內培養48h后,每孔加入5μlmts,繼續培養4h后,將96孔板置于酶標儀中,在490nm波長處測定吸光度值(od),計算細胞存活率,每個濃度平行6孔,實驗重復3次,計算公式如下:
藥理學數據:
表3燈盞乙素苷元及其衍生物對pkm2的抑制
表4燈盞乙素苷元及其衍生物體外抗腫瘤活性
以上數據表明,在sc13位上的o被s和se取代后,表現出對pkm2具有更強的抑制效果,相對于pkm1的抑制率,化合物sc4和sc6對pkm2表現出明顯的選擇性抑制。同時也表現出更好的抗腫瘤活性,特別是當o被se取代后,以上效果更為明顯。
當然,以上只是本發明的具體應用范例,本發明還有其他的實施方式,凡采用等同替換或等效變換形成的技術方案,均落在本發明所要求的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!