一種氨基酚基磷配體及其在乙烯聚合及齊聚中的應用的制作方法
技術領域:
本發明涉及催化劑技術領域,具體涉及一種氨基酚基磷配體及其在乙烯聚合及齊聚中的應用。
背景技術:
:
α-烯烴可以作為共聚單體用于線性低密度聚乙烯(lldpe)的生產以及用于生產合成潤滑油、原油減阻劑及表面活性劑等產品。通過乙烯與α-烯烴共聚生產的線性低密度乙烯是用途廣泛的合成樹脂,除了具有優良的韌性和抗環境應力開裂能力,耐腐蝕性、抗沖擊強度、撕裂強度和拉伸強度以外,還具有很好的加工性能,包括抗蠕變能力、脫模能力、成膜性、熱封性能等,因此被廣泛應用于制造醫療器械、薄膜,電纜的包覆料耐應力容器。常用的共聚單體有1-丁烯、1-己烯及1-辛烯等。有研究結果表明,以1-辛烯為共聚單體的得到的產品具有更好的性能。目前,乙烯四聚合成1-辛烯尚未實現大規模工業化生產,1-辛烯主要靠從非選擇性乙烯齊聚混合物中分離得到,因此成本較高。
自從sasol公司的研究人員首次報道由異丙胺合成的pnp配體與乙鉻鹽的配合物高效地催化乙烯選擇性四聚得到以1-辛烯為主要的齊聚產物以來,對于鉻系乙烯四聚催化劑的研究主要集中在配體的結構對催化效果的影響方面。盡管在催化活性等方面已取得了諸多喜人的成果,但對1-辛烯的選擇性遠未達到乙烯三聚工藝的水平,而新結構的配體的研發是解決這一問題的根本途徑。同時,新結構的配體及其配合物在乙烯的聚合及共聚合中的應用研究也是促進聚乙烯工業技術進步的重要推動力。
技術實現要素:
:
本發明的目的是為了克服上述現有技術存在的不足之處,而提供一種氨基酚基磷配體及其在乙烯聚合及齊聚中的應用,它提供了一種具有多個配位位點的磷配體,該配體可以與不同的過渡金屬的鹽以預制配合物或現場混合的方式形成主催化劑。
為了解決背景技術所存在的問題,本發明是采用如下技術方案:該配體是基于氨基酚的含磷的衍生物,其結構式如下:
其中:
氨基酚結構中,氨基處于酚羥基的鄰位、間位或對位;
r1為碳原子數不大于10的烷基、環烷基、烷氧基、芳基;
r2為碳原子數小于10的烷基、環烷基、芳基、烷氧基;
r3為碳原子數小于10的烷基、環烷基、芳基、烷氧基
所述的使用方法:將磷配體與過渡金屬化合物通過預制或現場配制形成的配合物或混合物,過渡金屬鹽的正離子包括cr(ii)、cr(iii)、ni(ii)、fe(ii)、co(iii)、pd(ii)、zr(ii)、zr(iv)、pt(ii),其中優選cr(ii)、cr(iii)、ni(ii)、fe(ii)pd(ii);過渡金屬鹽的負離子包括no3-、so42-、cl-、br-和乙酰丙酮負離子。
所述的配合物或混合物中磷配體與過渡金屬化合物的摩爾比為0.1:1~10:1。
所述的配合物或混合物作為主催化劑與助催化劑組成的的催化劑,助催化劑包括烷基鋁、鋁氧烷和氯化烷基鋁中的一種或多種的組合;主催化劑的金屬與助催化劑中的鋁的摩爾比為1:1~1:5000。
所述的烷基鋁包括三甲基鋁、三乙基鋁、三異丁基鋁和三正己基鋁中的一種或多種的組合;鋁氧烷包括甲基鋁氧烷、乙基鋁氧烷和異丁基鋁氧烷中的一種或多種的組合;氯化烷基鋁包括乙基倍半氯化鋁、一氯二乙基鋁、倍半一氯二乙基鋁和二氯化乙基鋁中的一種或多種的組合。
所述的催化劑在催化乙烯的聚合、共聚合及齊聚反應中的應用:與乙烯共聚合的單體包括c18以下的端烯、丙烯腈、丙烯酰胺、n,n-二甲基丙烯酰胺、丙烯酸甲酯、丙烯酸乙酯、丙烯酸正丁酯和甲基丙烯酸甲。
所述的在催化乙烯聚合、共聚合、齊聚過程中,聚合溫度為0~100℃,聚合壓力為0.1~0.5mpa。
本發明的有益效果是提供了一種多位點的磷配體,基于這種配體,通過改變與過渡金屬及助催化劑的配比,可以靈活地催化不同類型的乙烯的齊聚、聚合及共聚反應。
附圖說明:
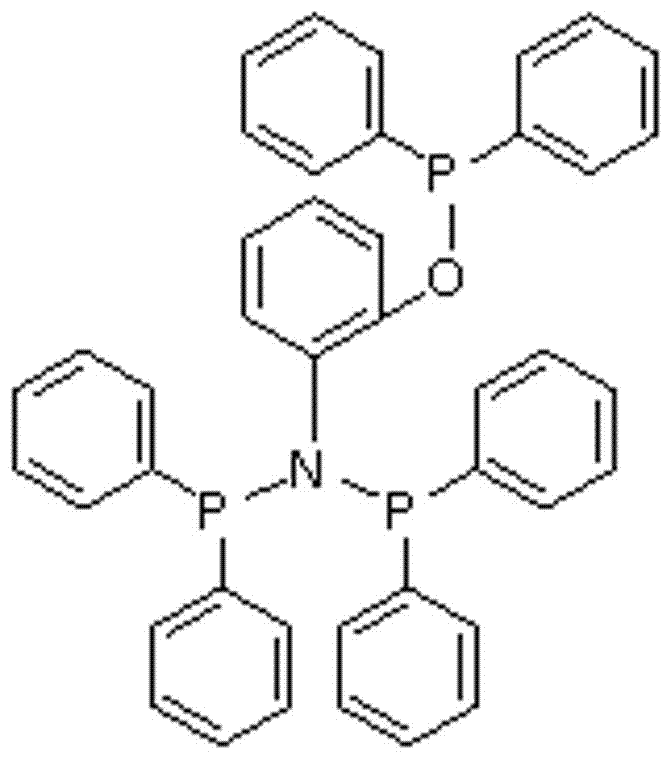
圖1是本發明所涉及配體的結構示意圖;
圖2是本發明配體1的結構示意圖;
圖3是本發明配體2的結構示意圖;
圖4是本發明配體3的結構示意圖;
圖5是本發明配體4的結構示意圖。
具體實施方式:
參照圖1,本發明具體采用如下實施方式:該配體是基于氨基酚的含磷的衍生物,其結構式如下:
其中:
氨基酚結構中,氨基處于酚羥基的鄰位、間位或對位;
r1為碳原子數不大于10的烷基、環烷基、烷氧基、芳基;
r2為碳原子數小于10的烷基、環烷基、芳基、烷氧基;
r3為碳原子數小于10的烷基、環烷基、芳基、烷氧基
所述的使用方法:將磷配體與過渡金屬化合物通過預制或現場配制形成的配合物或混合物,過渡金屬鹽的正離子包括cr(ii)、cr(iii)、ni(ii)、fe(ii)、co(iii)、pd(ii)、zr(ii)、zr(iv)、pt(ii),其中優選cr(ii)、cr(iii)、ni(ii)、fe(ii)pd(ii);過渡金屬鹽的負離子包括no3-、so42-、cl-、br-和乙酰丙酮負離子。
所述的配合物或混合物中磷配體與過渡金屬化合物的摩爾比為0.1:1~10:1,優選0.3:1~3:1。
所述的配合物或混合物作為主催化劑與助催化劑組成的的催化劑,助催化劑包括烷基鋁、鋁氧烷和氯化烷基鋁中的一種或多種的組合;主催化劑的金屬與助催化劑中的鋁的摩爾比為1:1~1:5000,優選1:100~1:1000。
所述的烷基鋁包括三甲基鋁、三乙基鋁、三異丁基鋁和三正己基鋁中的一種或多種的組合;鋁氧烷包括甲基鋁氧烷、乙基鋁氧烷和異丁基鋁氧烷中的一種或多種的組合;氯化烷基鋁包括乙基倍半氯化鋁、一氯二乙基鋁、倍半一氯二乙基鋁和二氯化乙基鋁中的一種或多種的組合。
所述的催化劑在催化乙烯的聚合、共聚合及齊聚反應中的應用:與乙烯共聚合的單體包括c18以下的端烯、丙烯腈、丙烯酰胺、n,n-二甲基丙烯酰胺、丙烯酸甲酯、丙烯酸乙酯、丙烯酸正丁酯和甲基丙烯酸甲。其中優選1-己烯、1-辛烯、1-癸烯、丙烯腈、丙烯酰胺、n,n-二甲基丙烯酰胺、丙烯酸甲酯、甲基丙烯酸甲酯。
所述的在催化乙烯聚合、共聚合、齊聚過程中,聚合溫度為0~100℃,聚合壓力為0.1~0.5mpa。
利用上述配體與過渡金屬(cr(ii)、cr(iii)、ni(ii)、fe(ii)、co(iii)、pd(ii)、zr(ii)、zr(iv)、pt(ii))的鹽按一定比例通過預制或現場配制形成的配合物或混合物,過渡金屬鹽的負離子包括no3-、so42-、cl-、br-和乙酰丙酮負離子,其中優選cl-和乙酰丙酮負離子。以制得的配合物或混合物作為主催化劑,在一定比例的助催化劑(包括烷基鋁、鋁氧烷和氯化烷基鋁中的一種或多種混合物)的作用下催化乙烯的齊聚、聚合和乙烯與極性及非極性單體的共聚反應。
利用氨基酚類化合物與取代的氯化磷作用,可以實現對氨基酚分子上的活潑氫的取代,從而形成具有多個配位位點的磷配體。這類磷配體可以與過渡金屬的化合物進行配位,在不同的配比下可以形成配合物或者混合物。這些配合物或混合物在烷基鋁及其衍生物的作用下,其金屬中心可以發生還原,從而具備了與乙烯類單體的配位能力,實現催化乙烯的齊聚、聚合及共聚等效果。
實施例1:
多位點磷配體1的制備
配體1的結構中,r1、r2、r3均為氫,氨基處于羥基的鄰位。其結構如圖2所示。
在高純氮氣保護下,在燒瓶中加入二苯基氯化磷5.8ml和40ml四氫呋喃,充分攪拌溶解后,加入三乙胺4.5ml,攪拌半小時后降溫至0℃,向溶液中逐滴滴加鄰氨基苯酚的溶液(1.24g鄰氨基苯酚溶于10ml四氫呋喃中),加完后繼續在0℃下攪拌半小時。撤冷自然升溫至室溫后繼續反應12小時。混合物過濾除去固體,所得濾液旋蒸去除溶劑得到粗產品。粗產品用無水乙醇重結晶并真空干燥后得到配體1,無色晶體,收率為81%。配體1元素分析c42h34nop3理論值(%):c,76.24;h,5.18;n,2.12;實測值(%):c,75.91;h,5.46;n,1.96。
實施例2:
配體1催化乙烯齊聚反應
將在80℃連續抽真空干燥2小時以上的500ml不銹鋼高壓反應釜用高純氮氣置換3次,然后再抽真空并用乙烯置換3次。在保持釜內微正壓的條件下向釜內注入150ml甲苯,開啟攪拌,加入溶有7.0mg乙酰丙酮鉻的甲苯溶液和溶有14.5mg配體1的甲苯溶液,5min后加入8.0ml助催化劑甲基鋁氧烷(10%的甲苯溶液)。在50℃下注入乙烯使釜內壓力達到并維持在3mpa,用質量流量計計量乙烯用量,并以700rpm的速度進行攪拌。反應30min后切斷乙烯進料,高壓釜降溫至0℃后泄壓至常壓,過濾去除催化劑與聚合物。濾液稱重并取樣用氣相色譜對其組份進行定量分析。催化活性為2.51×106gmolcr-1h-1,聚合物收率為1.9%,液相產物中1-己烯收率為14%,1-辛烯收率為73%。
實施例3:
多位點磷配體2的制備
配體2的結構中,r1為甲基,r2和r3均為氫,氨基處于羥基的間位。其結構如圖3所示。
配體2的合成過程與配體1相同,只不過氨基酚原料換成5-氨基鄰甲酚。粗產品用無水乙醇重結晶并真空干燥后得到配體2,無色晶體,收率為77%。1hnmr(δ,cdcl3,tms):2.10(s,3h),6.20(d,j=8.0,1h),6.55(s,1h),6.79(d,j=8.0,1h),6.98~7.76(m,30h)。
實施例4:
磷配體2的ni的配合物1的制備
無水無氧條件,室溫下取1.03g(1.52mmol)配體2加入到50ml正丁醇中攪拌溶解,再加入0.468g(1.52mmol)乙二醇二甲醚合二溴化鎳,升溫至55℃下攪拌反應24小時后撤去加熱,將反應器移入-15℃冰箱內靜置12小時析出棕黑色固體配合物。過濾后,濾餅用冷甲醇淋洗兩遍,真空干燥,得到產物1.10g(收率81%)。c43h36br2nniop3的元素分析理論值(%):c,57.76;h,4.06;n,1.57;實測值c,57.47;h,4.35;n,1.46.
實施例5:
配合物催化乙烯聚合反應
將在130℃連續干燥6小時以上的250m1玻璃三頸圓底燒瓶內置入磁力攪拌子并接裝schlenk標準真空-氣體置換系統及恒壓乙烯管線。加熱抽真空并用高純氮氣置換3次,然后再抽真空并用乙烯置換3次。向燒瓶內注入50m1的甲苯,然后加入溶有7.2mg配合物1的甲苯溶液,。按照預設的a1/ni比(500:1)加入10.0ml助催化劑乙基倍半氯化鋁(0.4mo1/l的己烷溶液),在20℃下,保持圓底瓶內latm的乙烯壓力用磁力攪拌器驅動劇烈攪拌,反應30min后切斷乙烯進料,用10%的鹽酸酸化乙醇溶液終止反應,過濾得到聚合物沉淀。聚合物用乙醇,水洗數次,真空烘干至恒重,稱量得聚合物16.7g。用聚合物的質量計算聚合活性為4.2×106g·mol-1(ni)·h-1,經測定聚合物mw=7.2×105g·mol-1,mw/mn=4.1。
實施例6
多位點磷配體3的制備
磷配體3的結構見圖4。
配體3的合成過程與配體1相同,只不過氨基酚原料換成4-氨基苯酚。粗產品用無水乙醇重結晶并真空干燥后得到配體3,無色晶體,收率為83%。配體3元素分析c42h34nop3理論值(%):c,76.24;h,5.18;n,2.12;實測值(%):c,75.87;h,5.39;n,1.85。
實施例7
配體3在乙烯共聚中的應用
將在130℃連續干燥6小時以上的250m1玻璃三頸圓底燒瓶內置入磁力攪拌子并接裝schlenk標準真空-氣體置換系統及恒壓乙烯管線。加熱抽真空并用高純氮氣置換3次,然后再抽真空并用乙烯置換3次。向燒瓶內注入100m1的甲苯,然后加入9.1ml(8.61g,0.1mol)丙烯酸甲酯攪拌均勻。向反應器中加入4.0ml甲基鋁氧烷溶液(10%的甲苯溶液),攪拌1小時。加入溶解6.2mg乙二醇二甲醚合溴化鎳的甲苯溶液和溶有8.8mg配體3的甲苯溶液。在30℃下,保持圓底瓶內latm的乙烯壓力用磁力攪拌器驅動劇烈攪拌,反應1h后切斷乙烯進料,用10%的鹽酸酸化乙醇溶液終止反應,過濾得到聚合物沉淀。聚合物用乙醇,水洗數次,真空烘干至恒重,稱量得聚合物10.4g。經測定聚合物mw=1.8×105g·mol-1。聚合物取樣溶于鄰二氯苯-d4,110℃下用核磁共振譜儀測定氫譜,根據氫譜數據計算極性單體丙烯酸甲酯的插入率為4.5%。
實施例8
多位點磷配體4的制備,配體4的結構如圖5所示。
配體4的合成過程與配體1相同,只不過氨基酚原料換成3-氨基苯酚,且氯化磷衍生物采用的是氯二(2-甲氧苯基)膦。粗產品用無水乙醇重結晶并真空干燥后得到配體4,無色晶體,收率為65%。配體3元素分析c48h46no7p3理論值(%):c,68.49;h,5.51;n,1.66;;實測值(%):c,68.15;h,5.63;n,1.75。
實施例9
配體4在乙烯共聚中的應用將在130℃連續干燥6小時以上的250m1玻璃三頸圓底燒瓶內置入磁力攪拌子并接裝schlenk標準真空-氣體置換系統及恒壓乙烯管線。加熱抽真空并用高純氮氣置換3次,然后再抽真空并用乙烯置換3次。向燒瓶內注入100m1的甲苯,然后加入15.6ml(11.2g,0.1mol)的1-辛烯攪拌均勻。向反應器中加入1.5ml助催化劑乙基倍半氯化鋁(0.4mo1/l的己烷溶液)和0.14ml甲基鋁氧烷溶液(10%的甲苯溶液),攪拌1小時。加入分散了1.5mg氯化亞鐵的甲苯懸濁液和溶有6.7mg配體3的甲苯溶液。在30℃下,保持圓底瓶內latm的乙烯壓力用磁力攪拌器驅動劇烈攪拌,反應1h后切斷乙烯進料,用10%的鹽酸酸化乙醇溶液終止反應,過濾得到聚合物沉淀。聚合物用乙醇,水洗數次,真空烘干至恒重,稱量得聚合物6.2g。經測定聚合物mw=3.5×105g·mol-1,mw/mn=5.3。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 一種制備2,3,4?三乙酰基?1?(2?氯?4?硝基?苯基)?alpha?L巖藻吡喃糖苷的方法與制造工藝
- 利用混酚制備有機磷配體并進一步制備己二腈的方法與制造工藝
- 一種左西孟旦藥物中間體對乙酰氨基苯丙酮的合成方法與制造工藝
- 一種柳氨酚藥物中間體N?對羥基苯基水楊酰胺的合成方法與制造工藝
- 一種間尼索地平藥物中間體乙酰乙酸異丁酯的合成方法與制造工藝
- 含有高劑量和低劑量藥物的組合的口腔崩解片組合物的制造方法與工藝
- 一種新型甲氨基阿維菌素水分散粒劑及其制備方法與制造工藝
- 一種藥物中間體鄰乙酰氨基酚的合成方法與制造工藝
- 一種3?氨基鄰苯二甲酸二甲酯的制備方法與制造工藝
- 三芳胺端基含量可調的主鏈含S,S?二氧?二苯并噻吩的聚合物及其制備方法與應用與制造工藝
- 一種制備5-乙酰乙酰氨基苯并咪唑酮工藝過程中大顆粒物分離裝置的制造方法
- 一種用于回收5-乙酰乙酰氨基苯并咪唑酮粉末的裝置的制造方法
- 一種用于制備5-乙酰乙酰氨基苯并咪唑酮的氯苯與水分離裝置的制造方法
- 對乙酰氨基酚的合成工藝的制作方法
- 聚谷氨酸修飾電極及其在檢測藥物對乙酰氨基酚含量中的應用
- 一種地西泮藥物中間體2-氯乙酰氨基-5-氯二苯酮的合成方法
- 一種氨基甲酰尿嘧啶衍生物及其應用
- 2-乙酰氨基-1,5,-脫水-2-脫氧-d-葡萄糖醇在制備抗水稻白葉枯病菌活性藥物中的應用
- 對乙酰氨基酚精制工段所產生的廢活性炭的再利用工藝的制作方法
- 使用其中進行了調控使賴氨酰-tRNA合成酶被表達或不被表達的細胞或球狀聚集的細胞 ...的制作方法
- 一種制備2,3,4?三乙酰基?1?(2?氯?4?硝基?苯基)?alpha?L巖藻吡喃糖苷的方法與制造工藝
- 利用混酚制備有機磷配體并進一步制備己二腈的方法與制造工藝
- 一種左西孟旦藥物中間體對乙酰氨基苯丙酮的合成方法與制造工藝
- 一種柳氨酚藥物中間體N?對羥基苯基水楊酰胺的合成方法與制造工藝
- 一種間尼索地平藥物中間體乙酰乙酸異丁酯的合成方法與制造工藝
- 含有高劑量和低劑量藥物的組合的口腔崩解片組合物的制造方法與工藝
- 一種新型甲氨基阿維菌素水分散粒劑及其制備方法與制造工藝
- 一種藥物中間體鄰乙酰氨基酚的合成方法與制造工藝
- 一種3?氨基鄰苯二甲酸二甲酯的制備方法與制造工藝
- 三芳胺端基含量可調的主鏈含S,S?二氧?二苯并噻吩的聚合物及其制備方法與應用與制造工藝
- 一種制備5-乙酰乙酰氨基苯并咪唑酮工藝過程中大顆粒物分離裝置的制造方法
- 一種用于回收5-乙酰乙酰氨基苯并咪唑酮粉末的裝置的制造方法
- 一種用于制備5-乙酰乙酰氨基苯并咪唑酮的氯苯與水分離裝置的制造方法
- 對乙酰氨基酚的合成工藝的制作方法
- 聚谷氨酸修飾電極及其在檢測藥物對乙酰氨基酚含量中的應用
- 一種地西泮藥物中間體2-氯乙酰氨基-5-氯二苯酮的合成方法
- 一種氨基甲酰尿嘧啶衍生物及其應用
- 2-乙酰氨基-1,5,-脫水-2-脫氧-d-葡萄糖醇在制備抗水稻白葉枯病菌活性藥物中的應用
- 對乙酰氨基酚精制工段所產生的廢活性炭的再利用工藝的制作方法
- 使用其中進行了調控使賴氨酰-tRNA合成酶被表達或不被表達的細胞或球狀聚集的細胞 ...的制作方法