一種官能化氮化硼納米片及其制備方法和應用與流程
本發明涉及一種官能化六方氮化硼納米片及其分散液以及制備方法和應用,屬于納米材料技術領域。
背景技術:
六方氮化硼納米片通常指厚度在100納米以下的薄片狀材料,具有優異的導熱性、絕緣性、阻隔性、耐高溫和耐化學穩定性,而且比表面積很高,在散熱、絕緣、介電、微電子、儲能、水處理、催化、生物醫藥等領域具有非常廣闊的應用前景。目前氮化硼納米片的制備方法通常包括剝離和合成兩種途徑。剝離途徑是以六方氮化硼微片為原料,主要有微機械剝離、球磨剝離、超聲剝離、高壓流動剝離、化學剝離等方法。novoselov等早期報道通過微機械剝離獲得了氮化硼納米片(p.natl.acad.sci.usa.2005,102,10451.),但產率很低、分離困難,不容易放大。liluhua等報道用苯甲酸芐酯為介質對氮化硼微片進行低速球磨15小時,結合超聲和離心制備了厚度小于4nm的氮化硼納米片(j.mater.chem.,2011,21,11862.),但苯甲酸芐酯成本高、且產率較低。魯福身等將氮化硼在二氯亞砜中進行超聲處理,通過靜置或離心獲得氮化硼納米片(cn105293453a,公開日2016年2月3日),但所用二氯亞砜毒性大、易揮發,且單純超聲剝離效率低。郝霄鵬等使用酸性高錳酸鉀或熔融混合堿對氮化硼微片進行化學剝離(cn103626141a,公開日2014年3月12日;cn102976295a,公開日2013年3月20日),但存在廢酸廢堿處理問題。氮化硼納米片的合成途徑包括化學氣相沉積、固相轉化等方法,存在成本高、制備效率低、在溶劑中分散性差等問題。
技術實現要素:
本發明的目的是開發一種新方法實現六方氮化硼納米片的低成本、高效率、綠色制備,并實現其表面或邊緣的官能化以提高其在溶劑和樹脂基體中的分散性,從而促進氮化硼納米片的規模化制備和實際應用。
為了達到上述目的,本發明采用了如下技術方案:
一種官能化六方氮化硼納米片,厚度小于等于100nm,優選小于等于10nm,所述氮化硼納米片的表面或邊緣含有醇羥基、烷基、羧基、醚、酮或酯有機官能團中的一種或多種。
一種官能化六方氮化硼納米片分散液,所述分散液中含有權利要求1所述的官能化氮化硼納米片,溶劑為水、醇類、酮類、酯類、醚類、酰胺類、砜類、芳香族化合物中的一種或多種組合。
一種官能化六方氮化硼納米片的制備方法,包括如下步驟:
步驟一,將六方氮化硼粉末與含羥基小分子有機固體按比例混合均勻,所述含羥基小分子有機固體包括但不限于單糖、二糖、多糖、糖醇、檸檬酸及其水合物、檸檬酸鹽及其水合物、抗壞血酸、抗壞血酸鹽、脂肪醇、芳香醇、苯酚類、水楊酸、水楊酸鹽、鞣酸及其鹽中的一種或多種組合,所述氮化硼粉末與含羥基有機固體質量比為1:0.1~1:100,優選為1:2~1:30;
步驟二,對混合物施加機械作用力并保持一段時間,使它們發生相互作用,所述施加機械作用力的方式包括但不限于球磨、砂磨、珠磨、振動磨、研磨、輥壓、密煉、氣流沖擊、螺桿共混中的一種或多種組合,所述機械作用力的保持時間總共為0.1-72小時,優選為4-48小時;
步驟三,利用水或有機溶劑通過洗滌和分離過程去除混合物中多余的有機固體,獲得濕態官能化氮化硼,將所述濕態官能化氮化硼進一步干燥和研磨可獲得干態官能化氮化硼。所述水或有機溶劑能夠溶解所述有機固體,洗滌、分離過程可重復1-5次,得到的洗滌液可以通過蒸發回收有機固體,并將所述有機固體重新利用;
步驟四,將所述濕態或干態官能化氮化硼分散在溶劑中,靜置或離心,取上清液,所述溶劑包括但不限于純水、酸性水、堿性水、醇類、酮類、酯類、醚類、酰胺類、砜類、芳香族化合物等。所述分散的方法包括但不限于超聲處理、機械攪拌、高速剪切、高壓均質處理、高速射流、微流體處理、膠體磨處理等一種或多種的組合。
步驟五,將所述上清液過濾或高速離心,收集固體物質并干燥,得到官能化氮化硼納米片,所述干燥方法包括鼓風干燥、真空干燥、冷凍干燥、噴霧干燥、超臨界干燥等。
一種官能化六方氮化硼納米片分散液的制備方法,所述分散液主要由上述官能化氮化硼納米片的制備方法中的步驟四直接得到或將步驟五制得的官能化氮化硼納米片再次分散在溶劑中制得。
一種官能化六方氮化硼納米片的應用,將前述的官能化氮化硼納米片制成宏觀材料,所述宏觀材料的形態包括薄膜、氣凝膠、水凝膠、纖維、泡沫,所述宏觀材料的制備方法包括過濾、涂覆、冷凍干燥、水熱組裝或紡絲。
進一步地,所述宏觀材料的基體為環氧樹脂、硅橡膠、熱塑性塑料、聚氨酯、硅油、導熱油、蠟質中的一種或多種。
本發明的原理在于:六方氮化硼微片與含羥基有機固體在機械作用下發生碰撞、粉碎,斷裂的氮化硼邊緣與有機固體的羥基發生力化學反應,使有機官能團接枝到氮化硼的邊緣,氮化硼微片還會在剪切作用下發生滑動而變薄,從而獲得官能化氮化硼。洗滌后所得官能化氮化硼在水或有機溶劑中通過超聲分散或機械作用,進一步剝離為官能化氮化硼納米片,形成穩定的分散液;收集分散液中的固體物質,可獲得官能化氮化硼納米片。
本發明具有以下有益效果:
(1)氮化硼與含羥基有機固體在機械作用力下會發生力化學反應,形成官能化氮化硼,所述官能化氮化硼在超聲或機械力作用下很容易剝離為官能化氮化硼納米片,其操作簡便、剝離效率高。所制備的納米片表面和邊緣含有大量的醇羥基、烷基、羧基、醚、酮、酯等官能團,在水和各種極性溶劑中的分散性大為提高,方便涂覆、印刷等應用。
(2)采用糖類、檸檬酸、抗壞血酸等含醇羥基生物質固體研磨氮化硼,所制備的氮化硼納米片表面含有糖、檸檬酸、抗壞血酸等生物分子,因此與生物體具有良好的相容性和界面作用;容易通過氫鍵作用和共價鍵合等相互作用負載藥物分子和靶向分子,在生物成像、載藥等領域具有良好的應用前景。
(3)采用含醇羥基有機固體尤其是蔗糖、葡萄糖、檸檬酸等低成本可再生綠色生物質通過機械作用剝離氮化硼,避免了強酸、強堿、強氧化劑的使用。洗滌液可以通過蒸發回收含羥基有機固體并重新利用,生產成本低。
(4)所制備的官能化氮化硼納米片與常見極性樹脂基體如環氧樹脂、聚氨酯、硅橡膠、熱塑性塑料、硅油、導熱油、蠟質等具有更強的相互作用,能夠降低氮化硼與樹脂基體間的界面熱阻,從而提高復合材料的導熱性能、力學性能,降低氮化硼的添加量。
附圖說明
附圖1為氮化硼原料(a)和實施例1制備的官能化氮化硼納米片(b)的紅外圖譜。
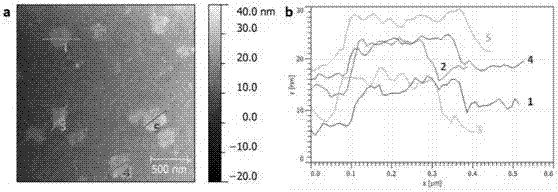
附圖2為實施例1制備的官能化氮化硼納米片的afm圖像(a)和典型高度曲線(b)。
附圖3為實施例2制備的官能化氮化硼納米片的afm圖像(a)和典型高度曲線(b)。
附圖4為氮化硼原料(a)和實施例3制備的官能化氮化硼納米片(b)的熱失重曲線。
附圖5為氮化硼原料(a)和實施例3制備的官能化氮化硼納米片(b)的b1sxps圖譜。
附圖6為實施例3制備的官能化氮化硼納米片的afm圖像(a)和典型高度曲線(b)。
附圖7為實施例4制備的官能化氮化硼納米片的sem圖像。
附圖8為實施例4制備的官能化氮化硼納米片的afm圖像(a)和典型高度曲線(b)。
附圖9為實施例5制備的薄膜的截面sem圖像,
附圖10為實施例5制備的氣凝膠的照片。
具體實施方式
下面結合具體實施例,進一步闡述本發明。應當理解,這些實施例僅用于說明本發明而不用于限制本發明的范圍。此外應理解,在閱讀了本發明的內容之后,本領域技術人員可以對本發明作各種改動或修改,這些等價形式同樣落于本申請所附權利要求書所限定的范圍。
在以下實施例中,六方氮化硼粉末購于秦皇島一諾高新材料開發有限公司,平均平面尺寸為30微米,其余試劑購自國藥集團。氮化硼納米片的形貌通過場發射掃描電子顯微鏡(sem,zeiss
實施例1
將10g蔗糖晶體與1g氮化硼粉末混合均勻,與110g直徑為6mm的不銹鋼球一起加入到250ml不銹鋼球磨罐中,密封后在qm-3sp2行星式球磨機上以200rpm的速度球磨24小時。打開球磨罐后聞到氨氣的氣味,說明氮化硼與蔗糖發生了力化學反應,其機理可能是硼原子與蔗糖羥基的氧結合,氮原子與羥基的氫結合,通過多步反應形成氨氣。對球磨產物用100ml去離子水超聲洗滌,用微孔濾膜過濾。其中所得濾液ph值為堿性,為溶解的氨或胺所致。將所得濾餅重復洗滌和過濾操作2次,每次用100ml去離子水。然后將濾餅在50ml去離子水和50ml乙醇混合溶劑中水浴超聲處理1小時,并在2000rpm下離心5分鐘,吸取上清液獲得官能化氮化硼納米片的水分散液。取適量該分散液進行過濾,并在60度下真空干燥12h,研磨后獲得官能化氮化硼納米片的粉末。
附圖1、附圖2分別為所得官能化氮化硼納米片的ftir圖譜和afm圖像。從ftir圖譜可見氮化硼納米片含有較強的o-h和c-h振動峰,說明其表面和邊緣接枝了較多的蔗糖分子,使其帶有大量的羥基。從afm圖像可見官能化氮化硼納米片的厚度約5~10nm,平面尺寸~300nm。
實施例2
將10g蔗糖晶體與1g氮化硼粉末混合均勻,與110g直徑為6mm的不銹鋼球一起加入到250ml不銹鋼球磨罐中,密封后在qm-3sp2行星式球磨機上以500rpm的速度球磨16小時。用100ml去離子水超聲洗滌,8000rpm離心10分鐘。將所得沉淀重復洗滌-離心操作2次,每次用100ml去離子水。然后將沉淀在60度下真空干燥12h,研磨后獲得官能化氮化硼納米片粉末。取0.2g粉末在20ml去離子水中水浴超聲分散1.5小時,靜置12小時后吸取上清液,獲得官能化氮化硼納米片的水分散液。附圖3為所得官能化氮化硼納米片的afm圖像,可見官能化氮化硼納米片的厚度約5nm。
實施例3
將10g蔗糖晶體與1g氮化硼粉末混合均勻,與100g直徑為10mm的不銹鋼球和30g直徑為1mm的不銹鋼球一起加入到250ml不銹鋼球磨罐中,密封后在qm-3sp2行星式球磨機上以500rpm的速度球磨24小時。
用100ml去離子水超聲洗滌,用微孔濾膜過濾。將所得濾餅重復洗滌-過濾操作3次,每次用100ml去離子水。然后取5g濕濾餅(固含量約10wt%)在50ml去離子水中水浴超聲分散2小時,在2000rpm的速度下離心10分鐘,吸取上清液獲得官能化氮化硼納米片的水分散液。取適量該分散液進行過濾,并在60度下真空干燥12h,研磨后獲得官能化氮化硼納米片的粉末。通過稱重法計算得知,洗滌純化的氮化硼通過一次超聲剝離和離心操作獲得氮化硼納米片的產率大于90%,分散液的濃度可以達到9.93mg/ml。
附圖4為所得官能化氮化硼納米片的熱失重曲線,可見官能化氮化硼納米片的失重率比氮化硼原料高出很多,說明其表面和邊緣接枝有較多的蔗糖分子。附圖5為氮化硼原料和官能化氮化硼納米片的b1sxps圖譜對比,可見球磨后b-n峰變弱,b-o峰明顯增強,說明有較多官能團鍵合到氮化硼納米片的表面或邊緣。附圖6為所得氮化硼納米片的afm圖像,可見納米片的厚度小于4nm。
實施例4
將20g檸檬酸一水合物與1g氮化硼粉末混合均勻,與200g直徑為6mm的不銹鋼球一起加入到250ml不銹鋼球磨罐中,密封后在qm-3sp2行星式球磨機上以500rpm的速度球磨24小時。
用100ml3wt%鹽酸溶液超聲洗滌10分鐘,并用微孔濾膜過濾。將所得濾餅用去離子水重復洗滌-過濾操作2次,每次用100ml去離子水。然后將濾餅在在60度下真空干燥12h,研磨后獲得官能化氮化硼的粉末。取0.2g粉末在10ml去離子水和10ml乙醇混合溶劑超聲分散2小時,在2000rpm的速度下離心15分鐘,吸取上清液獲得官能化氮化硼納米片的水分散液。取適量該分散液進行過濾,并在60度下真空干燥12h,研磨后獲得官能化氮化硼納米片的粉末。由稱重法計算得知,從洗滌所得官能化氮化硼通過一次超聲和離心獲得官能化氮化硼納米片的轉化率~66%。
附圖7為所得官能化氮化硼納米片的sem圖像,可見其平面尺寸~200nm。附圖8為所得氮化硼納米片的afm圖像,可見納米片的厚度小于5nm。
實施例5
將實施例3所得氮化硼納米片的分散液用微孔濾膜過濾,60℃真空干燥獲得氮化硼納米片的宏觀薄膜。附圖9為該薄膜的截面sem圖像,可見氮化硼納米片沿薄膜平面方向有序堆積。
將實施例3所得氮化硼納米片的分散液在玻璃容器中冷凍結冰,然后進行冷凍干燥12小時,獲得氮化硼納米片的氣凝膠。附圖10為該氣凝膠的照片。
實施例6
將1g正十八烷醇與1g氮化硼粉末混合均勻,然后密煉72h。對密煉產物利用乙醇洗滌,然后離心得到沉淀物,重復3次。然后將沉淀物分散于丙醇中,在高壓均質處理1h,靜置24h,取上清液,得到氮化硼納米片分散液。將所述分散液高速離心,收集固體物質,噴霧干燥,得到官能化氮化硼納米片。
實施例7
將30g麥芽糖與1g氮化硼粉末混合均勻,然后振動磨48h。對振動磨產物利用水超聲洗滌,然后離心得到沉淀物,重復5次。然后將沉淀物溶于乙酸乙酯中,再超聲處理1h,靜置24h,取上清液,得到氮化硼納米片分散液。將所述分散液高速離心,收集固體物質,真空干燥,得到官能化氮化硼納米片。
實施例8
實施例6所得官能化氮化硼納米片加入丙酮或四氫呋喃中,超聲分散均勻;然后加入環氧樹脂和固化劑混合均勻;最后加熱使丙酮揮發,并使樹脂固化,得到含有氮化硼納米片的導熱材料。
實施例9
將環氧樹脂和固化劑混合均勻,將實施例5所得的氮化硼納米片氣凝膠置于其中,在真空下進行填充和脫泡,然后加熱固化,得到含有官能化氮化硼氣凝膠的復合導熱材料。
實施例10
在實施例6所得到的官能化氮化硼納米片粉末加入適量導熱油或冷凍液中,高速機械攪拌使之混合均勻,獲得改性導熱油或冷凍液。
- 還沒有人留言評論。精彩留言會獲得點贊!