前列腺癌診斷用基因組及其應用的制作方法
本發明涉及前列腺癌診斷用基因組及其應用,特別是涉及ar、hoxc6和nkx2-2基因及其在制備或篩選診斷的藥物或試劑盒、或篩選治療前列腺癌的藥物中的應用。
背景技術:
惡性腫瘤是威脅人類健康的最危險因素之一,我國每年僅腫瘤死亡人數近200萬人,惡性腫瘤造成的經濟損失高達1400多億元人民幣。在我國,前列腺癌位于男性惡性腫瘤發病率第六位,在男性泌尿系腫瘤中排名第一位;而在美國前列腺癌發病率位居男性惡性腫瘤的第一位,死亡率僅次于肺癌位于第二位。雖然前列腺癌有比較好的預后,但對于男性來說仍然是造成死亡的重要癌癥。隨著男性防癌意識的增強和前列腺癌篩查方法的廣泛應用,早期發現的前列腺癌中,只有約5%初步診斷為無法治愈。然而30%-40%的前列腺癌患者最終會死于前列腺癌復發。
預后是指發病后,疾病未來過程的一種預先估計。在醫學上,“預后”是指根據經驗預測的疾病發展情況。預后主要涉及到三個方面,將發生什么結果、發生不良結果的可能性有多大、什么時候發生。預后分析是對疾病發病后發展為各種不同結局的預測;是臨床非常實用、對臨床很有指導作用的臨床研究。研究和評級預后的目的,為了便于了解各種疾病對人類危害性的大小、探索影響預后的因素、研究改善預后的具體措施。
腫瘤標識物是可以在血清、血漿、其他體液、組織提取物或石蠟固定的組織中檢測到的自然生產的分子。腫瘤標識物可以為前列腺癌的診斷,選擇治療方案、預測疾病進程和復發以及治療效果進行監測等提供有價值的信息。前列腺癌作為男性高發性惡性腫瘤,極為缺乏高效的腫瘤標記物。hpc1是最早被發現的高共顯前列腺癌易感基因,但估計在全部前列腺癌中僅占不到5%,其他已知的前列腺癌易感基因如msr1、hpc2等也僅能說明約1%左右的前列腺癌。目前我國在臨床上應用的前列腺癌標志物有psa、psca、psma等,在同一亞型中,不同病人的預后、存活時間、復發等都存在著顯著差異。這些數據表明,如何對前列腺癌進行準確的分型、盡早判斷腫瘤是否會復發、轉移,進而選擇合理的治療手段,提高前列腺癌患者的生存率和生活質量是前列腺癌治療的重要發展方向。
技術實現要素:
本發明的目的之一在于提供前列腺癌診斷用基因組ar、hoxc6和nkx2-2。
本發明的目的之二在于提供該基因組的篩選方法。
本發明的目的之三在于提供上述基因在制備篩選和診斷前列腺癌藥物中的應用。
本發明的目的之四在于提供上述基因制備篩選和診斷前列腺癌試劑盒中的應用。
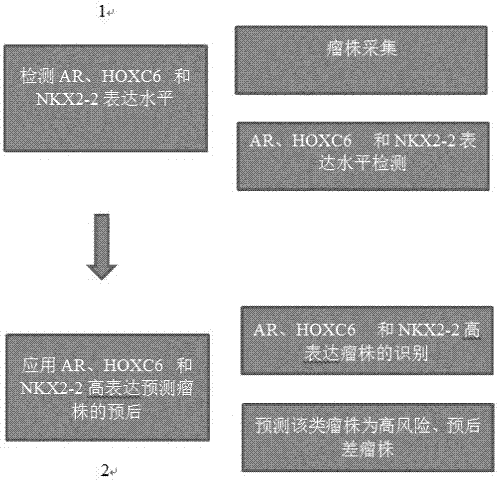
本發明發現ar、hoxc6和nkx2-2高表達與高風險前列腺癌的聯系,通過檢測前列腺癌樣株中ar、hoxc6和nkx2-2的表達水平,識別出ar、hoxc6和nkx2-2高表達的前列腺癌的樣株,并可進一步預測出該類前列腺癌可能具有較高的惡性程度,并表現出較差的預后。
為實現上述目的,本發明采用如下技術方案:
一種前列腺癌診斷用基因組,其特征在于該基因組為ar、hoxc6和nkx2-2基因。
一種前列腺癌的檢測方法,采用上述的前列腺癌診斷用基因,其特征在于該方法的具體步驟為:
a.選取前列腺癌樣本株組成3套數據;
b.采用simba算法尋找步驟a所得前列腺癌樣本株中的互斥轉錄因子;
c.采用費舍爾精確檢驗推測轉錄因子和其配對基因的調節關系;
d.利用最小描述方法,即兩個基因相關性最小情況,推測轉錄因子和相關的mirnas的調節網絡;
e.通過qrt-pcr檢測ar、hoxc6、nkx2-2的過表達。
上述的步驟b的具體步驟為:在r中調取simba程序包,對前列腺癌基因表達數據進行計算獲得互斥轉錄因子。
上述的步驟c的具體步驟為:在r中調用simba程序包中的函數init_network對上述找到的互斥轉錄因子搜尋靶標并建立初步網絡。
上述的步驟d的具體步驟為:在r中調用simba程序包中的函數filt_network對上一步構建的網絡進行優化,獲得最優化的mrna-mirna網絡。
上述的步驟e的具體步驟為:提取前列腺癌細胞株totalrna,按照試劑盒方法反轉錄后做qrt-pcr實驗檢測ar、hoxc6、nkx2-2的表達。
一種上述的前列腺癌診斷用基因組在制備篩選和診斷前列腺癌藥物中的應用。
一種上述的前列腺癌診斷用基因組在制備篩選和診斷前列腺癌試劑盒中的應用。
本發明的前列腺癌診斷用基因組通過下述方法來篩選:
a.選取前列腺癌樣本株組成3套數據;
b.采用simba算法尋找步驟a所得前列腺癌樣本株中的互斥轉錄因子;
c.采用費舍爾精確檢驗推測轉錄因子和其配對基因的調節關系;
d.利用最小描述方法,即兩個基因相關性最小情況,推測轉錄因子和相關的mirnas的調節網絡;
所述篩選診斷前列腺癌藥物具體指使用ar、hoxc6和nkx2-2,通過監測ar、hoxc6和nkx2-2表達量對可能作為診斷前列腺癌藥物使用的物質進行生物活性、藥理作用或要用價值的評估。
所述篩選診斷前列腺癌的試劑盒具體指使用ar、hoxc6和nkx2-2,通過監測ar、hoxc6和nkx2-2表達量對可能作為診斷前列腺癌的試劑盒進行性能的評估,所述性能如準確性、特異性、穩定性等。
所述篩選治療前列腺癌藥物具體指使用具體指使用ar、hoxc6和nkx2-2,通過監測ar、hoxc6和nkx2-2表達量對可能作為治療前列腺癌藥物使用的物質進行生物活性、藥理作用或藥用價值的評估。
優選的,所述制備診斷前列腺癌的藥物或試劑盒中,所述診斷前列腺癌具體指對前列腺癌惡性程度的預測和/或對前列腺癌預后的判斷。
更優選的,所述前列腺癌惡性程度的預測具體指通過具體指通過ar、hoxc6和nkx2-2表達量的檢測對前列腺癌樣株的惡性程度進行預測。
更優選的,所述前列腺癌預后的判斷具體指通過ar、hoxc6和nkx2-2表達量的檢測對前列腺癌病人的病程和/或結局進行預后判斷。
具體的,ar、hoxc6和nkx2-2高表達表示前列腺癌樣株具有較高的惡性程度,并表現出較差的預后;ar、hoxc6和nkx2-2低表達表示前列腺癌樣株具有較低的惡性程度,并表現出較好的預后。
本發明第二方面提供一種基于ar、hoxc6和nkx2-2高表達的高風險前列腺癌的新型診斷法,所述診斷方法具體為通過檢測腫瘤樣株中的ar、hoxc6和nkx2-2的表達水平對前列腺癌病人的預后進行判斷。
具體的,所述檢測腫瘤樣株中的ar、hoxc6和nkx2-2的表達水平是指將現有的數據庫中的腫瘤樣株的ar、hoxc6和nkx2-2表達量信息進行數據分布分析,再檢測患者前列腺癌樣株中ar、hoxc6和nkx2-2的表達水平,根據數據分布識別出這三個基因互斥表達的前列腺癌樣株。
在本發明實施例中,在統計學軟件r工作平臺中調用程序包rpackagelimma進行數據分布分析。
所述前列腺癌病人的預后進行判斷具體指,ar、hoxc6和nkx2-2高表達的這類前列腺癌株可能具有較高的惡性程度,并表現出較差的預后。
本發明發明人發現通過檢測瘤株中ar、hoxc6和nkx2-2的表達水平,能夠識別出ar、hoxc6和nkx2-2高表達的瘤株,并可進一步預測該類瘤株為高風險、預后差的類型。此外,經公開可得知前列腺癌病人chip-seq、mrna和mirna-seq的基因表達數據和存活數據證實,通過ar、hoxc6和nkx2-2表達量的表征,能夠有效預測出高風險、預后差的瘤株,可見ar、hoxc6和nkx2-2在制備或篩選診斷前列腺癌的藥物或試劑盒、或篩選治療前列腺癌的藥物的用途中具有高度產業利用價值。
附圖說明
圖1顯示為本發明的基于ar、hoxc6和nkx2-2高表達的高風險前列腺癌預后預測示意圖;
圖2顯示為互斥轉錄因子的算法流程;
圖3顯示為通過算法所構建的轉錄因子與mirnas的調解網絡圖;
圖4顯示為公開可得的不同癌癥或組織表達數據庫中ar、hoxc6和nkx2-2互斥高表達分布圖(a位tgca、b為robinson數據集,c為beltran數據集);
圖5顯示為ar、hoxc6和nkx2-2高表達的前列腺癌株的遷移與侵襲圖;
圖6顯示為ar、hoxc6和nkx2-2高表達的四套前列腺癌株的存活分析曲線圖(a為gse21032、b為glinskydataset、c為gse16560、d為tcgadatasets)。
具體實施方式
實施例一:
a.選取前列腺癌樣本株組成3套數據;
b.采用simba算法尋找步驟a所得前列腺癌樣本株中的互斥轉錄因子;
c.采用費舍爾精確檢驗推測轉錄因子和其配對基因的調節關系;
d.利用最小描述方法,即兩個基因相關性最小情況,推測轉錄因子和相關的mirnas的調解網絡;
e.從3套數據中都獲得ar、hoxc6和nkx2-2過表達;
實施例二:
a.選取前列腺癌樣本株組成3套數據;
b.采用simba算法尋找步驟a所得前列腺癌樣本株中的互斥轉錄因子;具體步驟為:在r中調取simba程序包,對前列腺癌基因表達數據進行計算獲得互斥轉錄因子。
c.采用費舍爾精確檢驗推測轉錄因子和其配對基因的調節關系;具體步驟為:在r中調用simba程序包中的函數init_network對上述找到的互斥轉錄因子搜尋靶標并建立初步網絡。
d.利用最小描述方法,即兩個基因相關性最小情況,推測轉錄因子和相關的mirnas的調解網絡;具體步驟為:在r中調用simba程序包中的函數filt_network對上一步構建的網絡進行優化,獲得最優化的mrna-mirna網絡。
實施例中所使用的前列腺癌樣株信息取自ncbigeo,并采用兩條標準對數據庫進行數據篩選:1,瘤株樣本數大于140例;2,具備臨床信息特別是病人隨訪的存活時間數據,因此得到3套數據,具體如下:
1)seriesaccession:gse21034id:200021034samples(370)
2)seriesaccession:gse21035id:200021035samples(231)
3)seriesaccession:gse21036id:200021036samples(142)
數據處理:
我們首先運用simba算法,參見中國專利cn105512509a去尋找前列腺癌樣株中的互斥轉錄因子,流程如圖2示。首先參見a圖,應用計算機二進制呈現出高斯混合分布離散連續的mrna和mirna表達譜成雙峰分布,找出異常表達的基因;由于原發癌在轉化成惡性癌癥的過程中,其驅動的轉錄因子和靶mirnas的表達是異常的,為此用b圖中費舍爾精確檢驗(fisher’sexacttests)推測轉錄因子和配對基因的調節關系;最后c圖中利用最小描述方法(mdl)也就是兩個基因相關性最小情況來推測轉錄因子和相關的mirnas的調解網絡,驅動性轉錄因子比非驅動性轉錄因子可以調節更多的mirnas。
利用這個計算方法,我們重構了前面的三套前列腺癌基因表達數據庫中轉錄因子的調節網絡,如圖3所示我們發現了三個互斥高表達的轉錄因子ar、hoxc6和nkx2-2及與之相關的mirnas,這些mirnas數量約占所有mirnas的數量的20%。隨后我們通過qrt-pcr檢測ar、hoxc6、nkx2-2的過表達,然后使用過表達的數據去檢測癌細胞轉移性的病人。
同時分析三套樣本獨立數據發現ar、hoxc6和nkx2-2互斥高表達在前列腺癌病人中是高度保守的,結果如圖4所示,其中,a圖數據庫來源tgca;b圖數據來源robinson數據集;c圖數據來源beltran數據集。虛線是轉錄因子高表達的中斷線。
同時如圖5所示,高表達ar、hoxc6和nkx2-2后細胞的遷移與侵襲能力也增強了。相對于無ar、hoxc6和nkx2-21組而言,ar、hoxc6和nkx2-2高表達組前列腺癌樣株均具有較高的遷移與侵襲能力。
依據瘤株樣本的存活數據進行存活曲線分析,結果如圖6所示。其中,橫坐標表示的是瘤株樣本來源的病人存活時間,縱坐標表示的是瘤株樣本來源的病人前列腺癌復發幾率(無轉移生存率)。由圖6中各圖可見,相對于ar、hoxc6和nkx2-21低表達組而言(黑色曲線所示),ar、hoxc6和nkx2-2高表達表示前列腺癌樣株均具有較高的惡性程度,并表現出較差的預后(淺藍色、紫色、深藍色和黃色曲線所示)。
綜上所述,本發明發明人發現通過檢測瘤株中ar、hoxc6和nkx2-2的表達水平,識別出ar、hoxc6和nkx2-2高表達的瘤株,并可進一步預測該類瘤株為高風險、預后差的類型。此外,經公開可得的前列腺癌病人chip-seq、mrna和mirna-seq的基因表達數據和存活數據證實,本發明能夠有效預測出高風險、預后差的瘤株,具有高度產業利用價值。
上述實施例僅例示性說明本發明的原理及其功效,a.選取前列腺癌樣本株組成3套數據;b.采用simba算法尋找步驟a所得前列腺癌樣本株中的互斥轉錄因子;具體步驟為:在r中調取simba程序包,對前列腺癌基因表達數據進行計算獲得互斥轉錄因子。c.采用費舍爾精確檢驗推測轉錄因子和其配對基因的調節關系;具體步驟為:在r中調用simba程序包中的函數init_network對上述找到的互斥轉錄因子搜尋靶標并建立初步網絡。d.利用最小描述方法,即兩個基因相關性最小情況,推測轉錄因子和相關的mirnas的調節網絡;具體步驟為:在r中調用simba程序包中的函數filt_network對上一步構建的網絡進行優化,獲得最優化的mrna-mirna網絡。而非用于限制本發明。任何熟悉此技術的人士皆可在不違背本發明的精神及范疇下,對上述實施例進行修飾或改變。因此,舉凡所屬技術領域中具有通常知識者在未脫離本發明所揭示的精神與技術思想下所完成的一切等效修飾或改變,仍應由本發明的權利要求所涵蓋。
非另外說明,本發明中所公開的實驗方法、檢測方法、制備方法均采用本技術領域常規的分子生物學、生物化學、染色質結構和分析、分析化學、細胞培養、重組dna技術及相關領域的常規技術。這些技術在現有文獻中已有完善說明,具體可參見sambrook等molecularcloning:alaboratorymanual,secondedition,coldspringharborlaboratorypress,1989andthirdedition,2001;ausubel等,currentprotocolsinmolecularbiology,johnwiley&sons,newyork,1987andperiodicupdates;theseriesmethodsinenzymology,academicpress,sandiego;wolffe,chromatinstructureandfunction,thirdedition,academicpress,sandiego,1998;methodsinenzymology,vol.304,chromatin(p.m.wassarmananda.p.wolffe,eds.),academicpress,sandiego,1999;和methodsinmolecularbiology,vol.119,chromatinprotocols(p.b.becker,ed.)humanapress,totowa,1999等。
- 還沒有人留言評論。精彩留言會獲得點贊!