一種納米復合物及其制備方法與應用與流程
本發明屬于高分子化學與生物醫學工程領域,尤其涉及一種納米復合物及其制備方法與應用。
背景技術:
陽離子聚合物載體是非病毒載體中的重要一類,此類材料能夠通過靜電相互作用和帶負電荷的核酸分子相結合,確保核酸分子在輸送過程中的完整性。相對于病毒載體,非病毒載體的發展面對的一個主要挑戰就是:如何提高轉染效率。
溶酶體逃逸是基于陽離子載體的核酸輸送效率提高的一個關鍵點。一般情況下,如果細胞內吞的納米顆粒物轉運到溶酶體中,將面臨被溶酶體水解酶降解的危險。因此如果polyplexes細胞內化轉運到溶酶體中時,必須設法突破溶酶體膜的屏障,盡早地實現溶酶體逃逸。當前陽離子聚合物核酸載體材料聚乙烯胺(pei)被公認為基因轉染的“黃金標準”,主要是由于pei具有較強的“質子海綿”效應,能夠有效的實現溶酶體逃逸,獲得較高的轉染水平。在生理ph7.4條件下,pei只有15~20%的氨基可以被質子化;但在溶酶體通路中,隨著ph的降低,pei上氨基的質子化程度顯著提高,導致納米復合物的表面正電荷密度上升,對溶酶體膜的擾動作用增強。同時,由于質子化過程引起大量質子和氯離子的流入,提高了溶酶體內的滲透壓,進一步降低了溶酶體膜的穩定性,有助于納米復合物實現溶酶體逃逸和轉運細胞的表達效率。
除了質子海綿效應外,光化學內化(photochemicalinternalization,pci)技術也是一個輔助大分子及納米粒子實現溶酶體逃逸的有效策略。pci技術基于光動力學治療(photodynamictherapy,pdt)的原理,通過使用光、氧氣和光敏劑相互作用促進大分子及納米粒子的溶酶體逃逸。通常,pci策略是通過納米復合物體系中引入卟啉、酞菁等具有強光誘導單線態氧產生能力的光敏劑,從而增強溶酶體膜的通透性,提高納米粒子的細胞質釋放率。
共軛聚電解質材料(conjugatedpolyelectrolyte,cpe)是一類具有由不飽和鍵和單鍵交替排列而成的主鏈和離子化側鏈的聚合物,同時具備獨特的光學性質及良好的親水性,在生物醫學領域受到研究者們的青睞。例如,陽離子聚噻吩(cationicpolythiophene,cpt)是一類易于制備、生物兼容性良好、光學性質優異的共軛聚電解質材料,在細胞分化、生物傳感器、熒光探針、熒光成像、仿生人造器官等生物領域具有廣泛的應用價值。類似的,陽離子聚苯撐乙炔(cationicpoly(phenyleneethynylene),cppe)也被應用于生物傳感、熒光成像、抗菌等生物醫學領域。cpe一般具有對氧分子的光敏化能力。
通常,陽離子聚合物載體材料往往需要相對較高的核酸用量以及較多的聚合物過量使用(96孔板中,劑量一般為5μg/ml或以上,n/p一般為10或以上;n/p越高,意味著載體材料用量越大),既不經濟,也不安全。眾所周知,使用過多的載體材料是引起細胞毒性最主要的原因。但是在陽離子聚合物載體體系中存在的普遍問題是,一旦降低聚合物和核酸分子的用量,就會導致核酸輸送效率嚴重偏低的不利結果。
因此,在聚合物載體材料和核酸分子用量雙降的前提下,采取新的聚合物載體體系構建策略,開發核酸輸送的有效增強方法,將使聚合物載體材料在生物醫學應用領域具有更廣泛的應用價值。
技術實現要素:
發明目的:本發明的第一目的是提供一種能夠在低質粒和聚合物用量的條件下有效增強核酸轉運效率的納米復合物;
本發明的第二目的是提供該復合物的制備方法;
本發明的第三目的是提供該復合物的應用。
技術方案:本發明的納米復合物,包括:陽離子聚合物載體、核酸分子及共軛聚電解質;其中,共軛聚電解質及陽離子聚合物載體的正電荷總和與核酸分子的負電荷之比為1~20:1,共軛聚電解質與陽離子聚合物載體的正電荷之比為0.01~10%。
本發明添加微量共軛聚電解質,通過共軛聚電解質材料的光敏化產生ros的作用,增強溶酶體膜通透性的改變、輔助納米復合物的溶酶體逃逸。
進一步說,所述陽離子聚合物載體包括聚氨基酸衍生物、聚糖衍生物或聚酰胺衍生物。優選的,聚氨基酸衍生物為可降解陽離子星形聚天冬酰胺載體材料,其由聚天冬酰胺主鏈和二乙烯三胺側鏈構成,結構式如下所示:
再進一步說,所述共軛聚電解質包括陽離子聚噻吩、陽離子聚苯撐乙炔、陽離子聚芴、陽離子聚苯撐乙烯或陽離子聚電解質共聚物。優選的,陽離子聚噻吩為陽離子線形聚噻吩或陽離子支化聚噻吩。其中,陽離子線形聚噻吩的結構式為:
r為
陽離子支化聚噻吩的結構式為:
其中,m/n=1/10~1/100,n=10~250,
r為
優選的,陽離子聚苯撐乙炔的結構式為:
其中,n=10~250。
本發明制備納米復合材料的方法,包括如下步驟:將陽離子聚合物載體與核酸分子混合,加入共軛聚電解質,靜置10~60min,得到多組份納米復合物。
本發明所述的納米復合物應用于增強核酸的輸送。
有益效果:與現有技術相比,本發明的顯著優點為:通過在聚合物載體及核酸分子體系基礎上,引入微量共軛聚電解質材料,形成該納米復合物,采用非共價組裝途徑對納米復合物表面進行功能化改造,不僅穩定性強,且應用于核酸的輸送中,在光照或非光照條件下均能夠增強溶酶體膜的滲透性,幫助納米復合物實現溶酶體逃逸,不會對細胞造成顯著損傷,增強基因輸送及核酸轉運效率,滿足基因輸送安全高效的要求;同時,其制備方法設計合理,制備過程簡單,大幅減少載體材料的使用量,具有廣闊的應用前景。
附圖說明
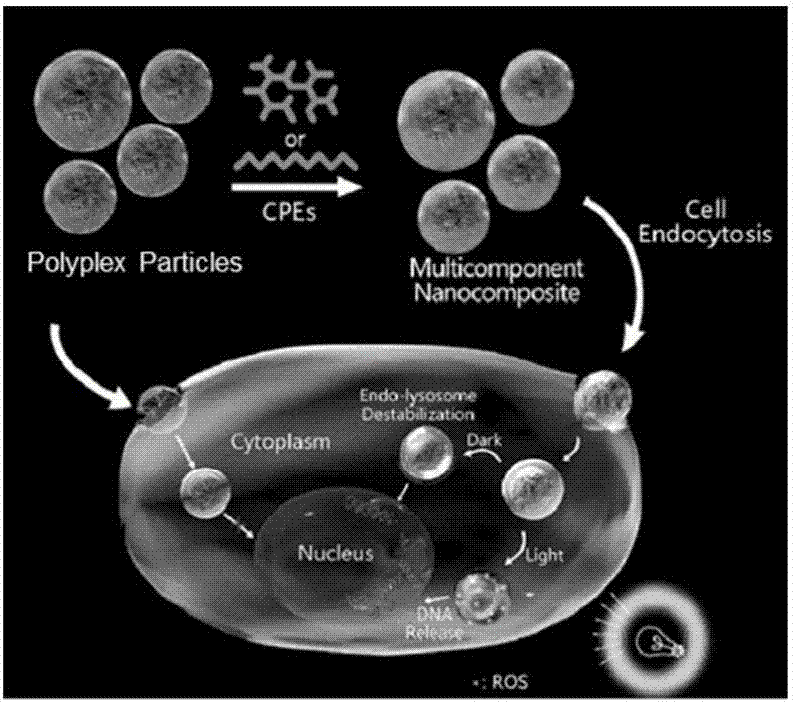
圖1為多組份納米復合物體系修飾增強核酸輸送的示意圖;
圖2a為陽離子線性聚噻吩的制備工藝流程圖;
圖2b為陽離子支化聚噻吩的制備工藝流程圖;
圖3a為lptm和bptm在cdcl3中的1hnmr表征圖;
圖3b為lpt和bpt在cdcl3中的1hnmr表征圖;
圖3c為脂溶性線性/支化聚噻吩的gpc表征,thf為流動相圖;
圖3d為脂溶性線性/支化聚噻吩由gpc測試結果計算得出的分子量和pdi圖;
圖4為陽離子聚噻吩構建的多組份納米復合物的尺寸和表面電勢圖;
圖5為陽離子聚噻吩構建的多組份納米復合物的eb競爭實驗圖;
圖6為陽離子線性/支化聚噻吩構建的多組份納米復合物對hepg2細胞作用24h的細胞毒性圖;
圖7a為無光照條件下,陽離子星形聚天冬酰胺載體材料(sp)在hela細胞內引起的光誘導ros檢測結果圖;
圖7b為以dcfh-da為ros熒光成像探針,光照條件下,陽離子星形聚天冬酰胺載體材料(sp)在hela細胞內引起的光誘導ros檢測結果圖;
圖8a為無光照條件下,聚乙烯胺載體材料(pei)在hela細胞內引起的光誘導ros檢測結果圖;
圖8b為以dcfh-da為ros熒光成像探針,光照條件下,聚乙烯胺載體材料(pei)在hela細胞內引起的光誘導ros檢測結果圖;
圖9a為無光照條件下,5%的陽離子支化聚噻吩(cbptm-1)構建的多組份納米復合物在hela細胞內引起的光誘導ros檢測結果圖;
圖9b為以dcfh-da為ros熒光成像探針,光照條件下,5%的陽離子支化聚噻吩(cbptm-1)構建的多組份納米復合物在hela細胞內引起的光誘導ros檢測結果圖;
圖10a為無光照條件下,2.5%的陽離子支化聚噻吩(cbptm-1)構建的多組份納米復合物在hela細胞內引起的光誘導ros檢測結果圖;
圖10b為以dcfh-da為ros熒光成像探針,光照條件下,2.5%的陽離子支化聚噻吩(cbptm-1)構建的多組份納米復合物在hela細胞內引起的光誘導ros檢測結果圖;
圖11a為無光照條件下,5%的陽離子線性聚噻吩(clptm)構建的多組份納米復合物在hela細胞內引起的光誘導ros檢測結果圖;
圖11b為以dcfh-da為ros熒光成像探針,光照條件下,5%的陽離子線性聚噻吩(clptm)構建的多組份納米復合物在hela細胞內引起的光誘導ros檢測結果圖;
圖12a為無光照條件下,2.5%的陽離子線性聚噻吩(clptm)構建的多組份納米復合物在hela細胞內引起的光誘導ros檢測結果圖;
圖12b為以dcfh-da為ros熒光成像探針,光照條件下,2.5%的陽離子線性聚噻吩(clptm)構建的多組份納米復合物在hela細胞內引起的光誘導ros檢測結果圖;
圖13a為無光照條件下,5%的陽離子線性聚噻吩(clpt-1)構建的多組份納米復合物在hela細胞內引起的光誘導ros檢測結果圖;
圖13b為以dcfh-da為ros熒光成像探針,光照條件下,5%的陽離子線性聚噻吩(clpt-1)構建的多組份納米復合物在hela細胞內引起的光誘導ros檢測結果圖;
圖14a為無光照條件下,2.5%的陽離子線性聚噻吩(clpt-1)構建的多組份納米復合物在hela細胞內引起的光誘導ros檢測結果圖;
圖14b為以dcfh-da為ros熒光成像探針,光照條件下,2.5%的陽離子線性聚噻吩(clpt-1)構建的多組份納米復合物在hela細胞內引起的光誘導ros檢測結果圖;
圖15a為無光照條件下,5%的陽離子支化聚噻吩(cbpt-1)構建的多組份納米復合物在hela細胞內引起的光誘導ros檢測結果圖;
圖15b為以dcfh-da為ros熒光成像探針,光照條件下,5%的陽離子支化聚噻吩(cbpt-1)構建的多組份納米復合物在hela細胞內引起的光誘導ros檢測結果圖;
圖16a為無光照條件下,2.5%的陽離子支化聚噻吩(cbpt-1)構建的多組份納米復合物在hela細胞內引起的光誘導ros檢測結果圖;
圖16b為以dcfh-da為ros熒光成像探針,光照條件下,2.5%的陽離子支化聚噻吩(cbpt-1)構建的多組份納米復合物在hela細胞內引起的光誘導ros檢測結果圖;
圖17a為無光照條件下,hela在無血清培養基中與聚乙烯胺載體材料(pei)共育5h后,在綠色通道的溶酶體通透性檢測結果圖;
圖17b為無光照條件下,hela在無血清培養基中與聚乙烯胺載體材料(pei)共育5h后,在紅色通道的溶酶體通透性檢測結果圖;
圖17c為無光照條件下,hela在無血清培養基中與聚乙烯胺載體材料(pei)共育5h后,在合并通道的溶酶體通透性檢測結果圖;
圖17d為光照條件下,hela在無血清培養基中與聚乙烯胺載體材料(pei)共育5h后,在綠色通道的溶酶體通透性檢測結果圖;
圖17e為光照條件下,hela在無血清培養基中與聚乙烯胺載體材料(pei)共育5h后,在紅色通道的溶酶體通透性檢測結果圖;
圖17f為光照條件下,hela在無血清培養基中與聚乙烯胺載體材料(pei)共育5h后,在合并通道的溶酶體通透性檢測結果圖;
圖18a為無光照條件下,hela在無血清培養基中與陽離子星形聚天冬酰胺載體材料(sp)共育5h后,在綠色通道的溶酶體通透性檢測結果圖;
圖18b為無光照條件下,hela在無血清培養基中與陽離子星形聚天冬酰胺載體材料(sp)共育5h后,在紅色通道的溶酶體通透性檢測結果圖;
圖18c為無光照條件下,hela在無血清培養基中與陽離子星形聚天冬酰胺載體材料(sp)共育5h后,在合并通道的溶酶體通透性檢測結果圖;
圖18d為光照條件下,hela在無血清培養基中與陽離子星形聚天冬酰胺載體材料(sp)共育5h后,在綠色通道的溶酶體通透性檢測結果圖;
圖18e為光照條件下,hela在無血清培養基中與陽離子星形聚天冬酰胺載體材料(sp)共育5h后,在紅色通道的溶酶體通透性檢測結果圖;
圖18f為光照條件下,hela在無血清培養基中與陽離子星形聚天冬酰胺載體材料(sp)共育5h后,在合并通道的溶酶體通透性檢測結果圖;
圖19a為無光照條件下,hela在無血清培養基中與陽離子線性聚噻吩(clpt-2)共育5h后,在綠色通道的溶酶體通透性檢測結果圖;
圖19b為無光照條件下,hela在無血清培養基中與陽離子線性聚噻吩(clpt-2)共育5h后,在紅色通道的溶酶體通透性檢測結果圖;
圖19c為無光照條件下,hela在無血清培養基中與陽離子線性聚噻吩(clpt-2)共育5h后,在合并通道的溶酶體通透性檢測結果圖;
圖19d為光照條件下,hela在無血清培養基中與陽離子線性聚噻吩(clpt-2)共育5h后,在綠色通道的溶酶體通透性檢測結果圖;
圖19e為光照條件下,hela在無血清培養基中與陽離子線性聚噻吩(clpt-2)共育5h后,在紅色通道的溶酶體通透性檢測結果圖;
圖19f為無光照條件下,hela在無血清培養基中與陽離子線性聚噻吩(clpt-2)共育5h后,在合并通道的溶酶體通透性檢測結果圖;
圖20為陽離子線性/支化聚噻吩與sp構建的多組份納米復合物對hela細胞轉染表達luciferase的質粒pluci40h的轉染情況圖;
圖21a為bpei在無光照條件下對hela細胞轉染表達綠色熒光蛋白的質粒pgfp40h的轉染情況圖;
圖21b為bpei在光照條件下對hela細胞轉染表達綠色熒光蛋白的質粒pgfp40h的轉染情況圖;
圖22a為sp在無光照條件下對hela細胞轉染表達綠色熒光蛋白的質粒pgfp40h的轉染情況圖;
圖22b為sp在光照條件下對hela細胞轉染表達綠色熒光蛋白的質粒pgfp40h的轉染情況圖;
圖23a為0.1%的陽離子聚苯撐乙炔(pim-1)構建的多組份納米復合物在無光照條件下對hela細胞轉染表達綠色熒光蛋白的質粒pgfp40h的轉染情況圖;
圖23b為0.1%的陽離子聚苯撐乙炔(pim-1)構建的多組份納米復合物在光照條件下對hela細胞轉染表達綠色熒光蛋白的質粒pgfp40h的轉染情況圖;
圖24a為0.25%的陽離子聚苯撐乙炔(pim-1)構建的多組份納米復合物在無光照條件下對hela細胞轉染表達綠色熒光蛋白的質粒pgfp40h的轉染情況圖;
圖24b為0.25%的陽離子聚苯撐乙炔(pim-1)構建的多組份納米復合物在光照條件下對hela細胞轉染表達綠色熒光蛋白的質粒pgfp40h的轉染情況圖;
圖25a為0.5%的陽離子聚苯撐乙炔(pim-1)構建的多組份納米復合物在無光照條件下對hela細胞轉染表達綠色熒光蛋白的質粒pgfp40h的轉染情況圖;
圖25b為0.5%的陽離子聚苯撐乙炔(pim-1)構建的多組份納米復合物在光照條件下對hela細胞轉染表達綠色熒光蛋白的質粒pgfp40h的轉染情況圖;
圖26a為1%的陽離子聚苯撐乙炔(pim-1)構建的多組份納米復合物在無光照條件下對hela細胞轉染表達綠色熒光蛋白的質粒pgfp40h的轉染情況圖;
圖26b為1%的陽離子聚苯撐乙炔(pim-1)構建的多組份納米復合物在光照條件下對hela細胞轉染表達綠色熒光蛋白的質粒pgfp40h的轉染情況圖;
圖27為無光照條件下,陽離子聚苯撐乙炔(pim-1、pim-2)與sp構建的多組份納米復合物對hela細胞轉染表達luciferase的質粒pluci40h的轉染情況圖,bpei和sp的轉染情況作為對照。
具體實施方式
下面通過實施例及附圖對本發明的技術方案進行詳細說明。
本發明的納米復合物,包括:陽離子聚合物載體、核酸分子及共軛聚電解質;其中,共軛聚電解質及陽離子聚合物載體的正電荷總和與核酸分子的負電荷之比為1~20:1,共軛聚電解質與陽離子聚合物載體的正電荷之比為0.01~10%。
其中,陽離子聚合物載體包括聚氨基酸衍生物、聚糖衍生物或聚酰胺衍生物等。具體而言,陽離子聚合物載體包括骨架及側鏈,其中,陽離子聚合物載體的骨架包括聚氨基酸、聚丙烯酰胺、聚甲基丙烯酰胺、聚丙烯酸酯、聚甲基丙烯酸酯、聚降冰片烯、聚乙烯胺、聚糖、聚酰胺、或基于上述骨架結構的共聚物等;陽離子聚合物側鏈基團包括烷基胺、烷基季銨鹽、烷基咪唑等,其中烷基可以是c1~c18鏈,烷基鏈上含有0~6個雜原子,聚合物分子量為2000~150000da。
共軛聚電解質包括陽離子聚噻吩、陽離子聚苯撐乙炔、陽離子聚芴、陽離子聚苯撐乙烯或陽離子聚電解質共聚物。具體而言,共軛聚電解質包括骨架及側鏈,其中,共軛聚電解質骨架包括聚噻吩、聚苯撐乙炔、聚芴、聚苯撐乙烯、聚丁二炔、或基于上述骨架結構的共聚物等;共軛聚電解質側鏈基團包括烷基胺、烷基季銨鹽、烷基咪唑等,其中烷基可以是c1~c18鏈,鏈上含有0~6個雜原子。聚合物分子量為1000~50000da。優選的,陽離子聚噻吩可包括線形陽離子聚噻吩或陽離子支化聚噻吩;陽離子聚苯撐乙炔可包括線形陽離子聚苯撐乙炔或陽離子支化聚苯撐乙炔;陽離子聚芴可為線形陽離子聚芴;陽離子聚苯撐乙烯可為線形陽離子聚苯撐乙烯;陽離子聚電解質共聚物可包括陽離子聚(苯撐乙炔-co-苯)、陽離子聚(芴-co-苯)等。
進一步說,本發明陽離子線性聚噻吩由如下步驟制備而得:
(1)分別催化氧化3-噻吩乙酸乙酯或者3-噻吩丙二酸二乙酯單體聚合,隨后清洗使鐵離子完全除去,得到兩類脂溶性線性聚噻吩;
(2)將上述兩類脂溶性線性聚噻吩分別經分離純化、胺解、質子化及透析后,即得到陽離子線性聚噻吩。
本發明陽離子支化聚噻吩由如下步驟制備而得:
(1)按摩爾比0.5~1:1將2-噻吩乙酸與n-羥基琥珀酰亞胺反應生成2-噻吩乙酸活性酯;
(2)將2-噻吩乙酸活性酯與三(2-氨乙基)胺及三乙胺在ch2cl2中反應,制得如下結構式的t3,其中,2-噻吩乙酸活性酯、三(2-氨乙基)胺、三乙胺的摩爾比為0.1~1:0.03~0.3:1,
(3)分別催化氧化噻吩乙酸乙酯與t3或者噻吩丙二酸二乙酯與t3聚合,隨后清洗,使鐵離子完全除去得到兩類脂溶性支化聚噻吩,其中,噻吩乙酸乙酯與t3的摩爾比為10~100:1,噻吩丙二酸二乙酯與t3的摩爾比為10~100:1;
(4)將上述兩類脂溶性支化聚噻吩分別經分離純化、胺解、質子化及透析后,即得到陽離子支化聚噻吩。
本發明陽離子聚苯撐乙炔cppe-det由如下步驟制備而得:
(1)將2,5-二碘-1,4-二苯乙酸溶于無水乙醇中,加入二氯亞砜,制得2,5-二碘-1,4-二苯乙酸乙酯,其中,2,5-二碘-1,4-二苯乙酸與二氯亞砜的摩爾比為10~100:1;
(2)將2,5-二碘-1,4-二苯乙酸乙酯溶于無水四氫呋喃和三乙胺中,分別加入pd(pph3)4、cui及三甲基硅烷乙炔,隨后加入四丁基氟化銨,沉淀制得ppe-et,其中,2,5-二碘-1,4-二苯乙酸乙酯、pd(pph3)4、cui及三甲基硅烷乙炔的摩爾比為0.25~0.5:0.001~0.01:0.001~0.01:1;
(3)將上述制得的ppe-et溶于nmp,加入二乙基三胺的nmp溶液,透析后制得cppe-det。
本發明陽離子聚苯撐乙烯由如下步驟制備而得:
(1)將2,5-二碘-1,4-二苯乙酸溶于無水乙醇,加入二氯亞砜,制得2,5-二碘-1,4-二苯乙酸乙酯,其中,2,5-二碘-1,4-二苯乙酸與二氯亞砜的摩爾比為10~100:1;
(2)將2,5-二碘-1,4-二苯乙酸乙酯、1,4-苯二乙烯溶于無水二氧六環,加入三乙胺和pd/c,反應制得ppv-et,其中,2,5-二碘-1,4-二苯乙酸乙酯、1,4-苯二乙烯的摩爾比為0.8~1.25:1;
(3)將上述ppv-et溶于nmp,加入二乙基三胺的nmp溶液,即制得cppv-det。
本發明陽離子共聚物聚(芴-co-苯)由如下步驟制備而得:
將fl-et、1,4-二苯硼酸溶于甲苯和碳酸鉀水溶液中,隨后加入四(三苯基膦)鈀,反應后制得pfp-et,將該pfp-et溶于干燥的nmp,加入r-nh2的nmp溶液,反應后制得cpfp-r,其中,fl-et、1,4-二苯硼酸、四(三苯基膦)鈀的摩爾比為10~25:10~25:1。
下述實施例中,如無特殊說明,均為常規方法,所用的實驗材料,如無特殊說明,均自常規生化試劑商店購買得到的。下述實施例中所述x%的共軛聚電解質用量,如無特殊說明,均為摩爾量的百分含量。以下實施例中的定量實驗,均設置三次重復試驗,結果取平均值。
實施例1陽離子聚噻吩的制備
1、陽離子線性聚噻吩
本發明的陽離子線性聚噻吩的結構式為:
制備方法包括如下步驟:
(1)脂溶性線性聚噻吩(lpt或lptm)的合成
氮氣保護下,將無水三氯化鐵(1.05g,6.5mmol)溶于無水硝基甲烷(25ml),另在氮氣保護下,將3-噻吩乙酸乙酯(0.50g,3.34mmol)或者3-噻吩丙二酸二乙酯(0.80g,3.31mmol)溶解于無水chcl3(25ml);冰浴下,將fecl3的硝基甲烷溶液逐滴加入反應體系,體系加熱至35℃,避光攪拌反應2h,停止反應后,體系先用0.01m鹽酸洗一遍,再用含有三乙醇胺(5%,w/v)和檸檬酸鈉(36.7g/l)的混合溶液洗三次,最后用氟化銨(10%,w/v)洗一次,使鐵離子完全除去;有機相用無水硫酸鈉干燥過夜后蒸除溶劑,得到的固體用少量氯仿溶解,再用乙醚重沉淀,得到脂溶性線性聚噻吩的混合物,該混合物進一步分別用尺寸排阻色譜法(sec)分離,得到脂溶性線性聚噻吩(lpt1、lpt2、lptm)。
(2)陽離子線性聚噻吩(clpt或clptm)的合成
稱取上述脂溶性線性聚噻吩(0.50g)溶于nmp(10ml)中,冰箱內放置6h,使其充分溶解;氬氣保護下,另取二乙烯三胺det(50eq.)溶于n-甲基吡咯烷酮,即nmp(8ml)中,并用冰鹽浴冷卻,隨后,攪拌下,將已置換氬氣的脂溶性線性聚噻吩的nmp溶液逐滴加入至det的nmp溶液中,反應4h,反應停止后,將體系慢慢逐滴加入預冷的ph=1的鹽酸中,冰鹽浴下大力攪拌;將鹽酸質子化后得到澄清透明的液體轉入透析袋(根據gpc測試結果確定使用何種截留分子量的透析袋)中,先用ph=2的鹽酸溶液透析1d,再用ph=4的鹽酸溶液透析1d,最后用中性去離子水透析2d,透析完成后,取出透析袋中液體,用0.22μm濾器過濾后,凍干得到陽離子線性聚噻吩(clptm、clpt-1、clpt-2)。
上述反應的流程如圖2a所示。
2、陽離子支化聚噻吩
本發明的陽離子支化聚噻吩的結構式為:
制備方法包括如下步驟:
(1)化合物t3的合成
將2-噻吩乙酸(0.30g,2.1mmol)溶于二氯甲烷(20ml),加入1-(3-二甲氨基丙基)-3-乙基碳二亞胺鹽酸鹽(0.48g,2.52mmol)及n-羥基琥珀酰亞胺(0.29g,2.52mmol),室溫下反應過夜;水洗、無水硫酸鈉干燥后,旋蒸除去溶劑,將上述2-噻吩乙酸活性酯溶于二氯甲烷(8ml),加入三(2-氨乙基)胺(0.08g,0.55mmol)和三乙胺(0.067g,0.66mmol)并攪拌反應24h,反應結束后,反應體系用飽和氯化銨水溶液洗兩次,飽和食鹽水洗一次,無水硫酸鈉干燥過夜,產物濃縮后用硅膠柱分離純化,得到白色固體t3(0.18g);將t3進行性能檢測,可得到如下所述的產物表征:核磁氫譜(400mhz,cdcl3):δ(ppm)7.19(3h),6.96-6.90(m,6h),6.70(brs,3h),3.75(s,6h),3.19(q,6h),2.47(t,6h).核磁碳譜(75mhz,dmso-d6):δ(ppm)169.7,138.1,127.0,126.4,125.2,53.7,37.6,37.0.高分辨質譜:m/zc24h31n4o3s3([m+h]+)計算值519.1558,實測值519.1557。上述表征數據表明t3結構無誤。
(2)脂溶性支化聚噻吩(bpt或bptm)的合成。
氮氣保護下,將無水三氯化鐵(1.05g,6.5mmol)溶解于無水硝基甲烷(25ml);另在氮氣保護下,將3-噻吩乙酸乙酯(0.50g,3.3mmol)或者3-噻吩丙二酸二乙酯(0.80g,3.3mmol)和t3(0.07g,0.14mmol)溶解于無水chcl3(25ml),冰浴下,將fecl3的硝基甲烷溶液逐滴加入反應體系,體系升溫至35℃,避光攪拌反應2h;停止反應后,體系先用0.01m鹽酸洗一遍,再用含有三乙醇胺(5%,w/v)和檸檬酸鈉(36.7g/l)的混合溶液洗三次,最后用氟化銨(10%,w/v)洗一次,使鐵離子完全除去;有機相用無水硫酸鈉干燥過夜后蒸除溶劑,得到的固體用少量氯仿溶解,再用乙醚重沉淀,得到脂溶性線性聚噻吩的混合物,該混合物進一步用尺寸排阻色譜法分離,收集各部分產物并分別濃縮干燥,即得到脂溶性支化聚噻吩bpt-1、bpt-2、bpt-3、bptm-1、bptm-2。
上述lpt1、lpt2、lptm的1hnmr和gpc的表征結果,bpt或bptm的1hnmr和gpc的表征結果如圖3a至3d所示。根據lpt1、lpt2、lptm的1hnmr和gpc的表征結果可知,低場、中高場以及高場的共振峰對應核磁譜圖中噻吩單元的氫,以聚苯乙烯為參照物的gpc分析顯示mw分子量分別為33、9、4kda,說明為聚噻吩結構;根據bpt或bptm的1hnmr和gpc的表征結果可知,在核磁氫譜方面,bpt和bptm除了具有噻吩單元的共振峰,還具有支化單元t3的共振峰,在gpc方面,bpt和bptm都出現在比單體更短的保留時間,上述結果表明bpt和bptm為聚合產物,且t3結構被成功整合到聚噻吩中。
(3)陽離子支化聚噻吩(cbpt或cbptm)的合成
稱取上述脂溶性支化聚噻吩(0.50g)溶于nmp(10ml)中,冰箱內放置6h,使其充分溶解;氬氣保護下,另取det(50eq.)溶于nmp(8ml)中并用冰鹽浴冷卻,隨后,攪拌下,將已置換氬氣的脂溶性支化聚噻吩的nmp溶液逐滴加入至det的nmp溶液中,反應4h;反應停止后,將體系慢慢逐滴加入預冷的ph=1的鹽酸中,冰鹽浴下大力攪拌。將鹽酸質子化后得到澄清透明的液體轉入透析袋(根據gpc測試結果確定使用何種截留分子量的透析袋)中,先用ph=2的鹽酸溶液透析1d,再用ph=4的鹽酸溶液透析1d,最后用中性去離子水透析2d,透析完成后,取出透析袋中液體,用0.22μm濾器過濾后,凍干得到陽離子支化聚噻吩(cbptm-1、cbptm-2、cbpt-1、cbpt-2、cbpt-3)。其制備工藝流程圖如圖2b所示。
在制備陽離子支化聚噻吩時,步驟(1)中,將2-噻吩乙酸與n-羥基琥珀酰亞胺的摩爾比替換成0.5:1或1:1,其中,2-噻吩乙酸活性酯與三(2-氨乙基)胺及三乙胺的摩爾比為0.1:0.03:1、0.5:0.1:1或1:0.3:1,均可制得該結構的陽離子支化聚噻吩;步驟(2)中,將噻吩乙酸乙酯與t3,或噻吩丙二酸二乙酯與t3的摩爾比替換為10:1、50:1、100:1,均可制得該結構的陽離子支化聚噻吩。
實施例2陽離子線性/支化聚噻吩構建的多組份納米復合物
原料組成:該復合物包括可降解陽離子星形聚天冬酰胺載體、dna及陽離子線性/支化聚噻吩;其中,陽離子線性/支化聚噻吩及可降解陽離子星形聚天冬酰胺載體的正電荷總和與dna的負電荷之比為2.5:1,陽離子線性/支化聚噻吩與可降解陽離子星形聚天冬酰胺載體的正電荷之比為2.5%或5%。
制備方法:在聚合物材料所含的胺基(包括1~4級胺)數目與核酸分子所含磷酸酯鍵數目之比為5的條件下(即n/p=5,由于只有一半胺基在中性條件下發生了質子化,此時正負電荷比為2.5:1),首先將可降解陽離子星形聚天冬酰胺載體(sp)材料和質粒dna用超純水稀釋到相同的體積后,將質粒溶液緩慢加入到sp材料中,完全加完后靜置15min,使材料與質粒自組裝形成穩定的納米復合物。隨后,向納米復合物體系中緩慢加入等體積的陽離子線性/支化聚噻吩,完全加完后再靜置15min,使陽離子線性/支化聚噻吩結合在納米復合物表面,組裝形成穩定的多組份納米復合物。
引入2.5%、5%陽離子線性/支化聚噻吩構建的多組份納米復合物體系,測定其水合動力學直徑及表面電勢,結果如圖4所示,加入少量的陽離子線性/支化聚噻吩后,納米復合物的尺寸變小,表面電勢提高。
如圖1所示,核酸分子被穩定包裝于內核處的納米復合材料被細胞攝取后,其表面的共軛聚電解質在非光照下擾動溶酶體膜,或者在光照下通過光化學內化效應進一步增加溶酶體膜的通透性,從而顯著提高核酸顆粒溶酶體逃逸的成功率以及最終的轉染效率。
性能檢測1陽離子線性/支化聚噻吩構建的多組份納米復合物的穩定性檢測
溴化乙錠(ethidiumbromide,eb)競爭實驗用于檢測陽離子線性/支化聚噻吩構建的多組份納米復合物的穩定性。實驗中eb終濃度為5μg/ml,質粒dna終濃度為1.6μg/ml,去離子為溶劑。以n/p=0.25為變化當量,逐步加入材料(加入材料溶液的體積盡量少,以免引起質粒濃度較大的變化),混勻后靜置5min測定eb的熒光發射光譜強度變化,溶液隨著材料加入,體系相對熒光強度(relativefluorescenceintensity,rfi)可以按如下公式計算得出:
rfi=(fx-f0)/(fmax-f0)
其中,f0表示僅有eb時,體系熒光強度;fmax表示eb中加入質粒dna,穩定5min后的熒光強度;fx表示eb與質粒dna的溶液中加入載體材料,穩定5min后熒光強度。為了避免陽離子噻吩材料的干擾,選用激發光波長為550nm,收集最大發射波長600nm處的熒光變化。結果如圖5所示,引入2.5%或5%的cpt修飾sp/pgfp納米復合物,有效提高了納米復合物的穩定性,且相比sp/dna和bpei/dna,cpt/sp/dna多組分納米復合物具有更高的穩定性,有助于提高在輸送過程中載體材料對dna的保護能力。
性能檢測2微量陽離子線性/支化聚噻吩構建的多組份納米復合物的細胞毒性實驗
收集對數生長期hepg2細胞,調整細胞懸液濃度,以每孔大約1.6×104個/200μl的細胞濃度接種到96孔板,置于含有5%co2的37℃培養箱內生長12-24h。當細胞融合70-80%時,將培養基吸除,pbs洗細胞1-2遍,加入含有預設濃度測試樣品的完全培養基,繼續孵育24h。24h后,吸除含有樣品的培養基,pbs洗細胞1-2遍,每孔加入100μl含有mtt(0.5mg/ml)的完全培養基,培養箱內繼續孵育4h。4h后吸除培養基,pbs輕洗細胞1-2遍。加入150μldmso,搖床上輕搖20min,使生成的甲瓚晶體充分溶解。酶標儀分別測定各孔在570nm、720nm波長下的吸收值,記錄結果。其中,每孔所記錄的720nm波長下的吸光度值為扣除背景用,即最終計算用的吸光度值為od570減去od720后的數值。細胞存活率可以按照如下公式計算:
細胞存活率(%)=(樣品組平均吸光度值/對照組平均吸光度值)×100%
注意當檢測光毒性時,則在多組份納米復合物體系與細胞共孵育4-6h時引入光照條件即可,其余操作與未光照組一致。
結果如圖6所示,sp/pgfp復合物的細胞毒性和cpts構建的多組份納米復合物體系的細胞毒性基本相近,分子量較低的陽離子聚噻吩材料,在光照及非光照條件下均未產生明顯的細胞毒性。
性能檢測3微量陽離子線性/支化聚噻吩構建的多組份納米復合物在細胞內引起ros水平變化測定
細胞內的活性氧可以氧化無熒光的dcfh生成有熒光的dcf,通過檢測dcf的熒光強度可以推算出細胞內活性氧水平。將ros捕捉劑dcfh-da用dmso溶解制備成10mm溶液,使用時用pbs稀釋至終濃度為20μm。以96孔板為例:收集對數生長期的細胞,調整細胞懸液濃度,以每孔大約1.6×104個/200μl的細胞濃度接種到96孔板,置于含有5%co2的37℃培養箱內生長12-24h。當細胞融合70-80%時,將培養基吸除,pbs洗細胞1-2遍,加入含有載體材料/質粒的納米復合物的不完全培養基。吸除每孔中含有材料的培養基,pbs洗1-2遍,加入dcfh-da探針溶液(終濃度為20μm),37℃細胞培養箱內避光孵育20min。去除探針溶液,pbs洗2-3遍。洗滌完后每孔加入100μl完全培養基,熒光倒置顯微鏡下觀察記錄dcfh-da捕捉了ros后的氧化產物dcf的綠色熒光(ex=465/95nm,em=515-555nm)。
當檢測光照作用時,則在多組份納米復合物體系與細胞共孵育4-6h時引入光照條件即可,其余操作與未光照組一致。
通過圖7a、7b至圖16a、16b所示,陽離子線性/支化聚噻吩修飾形成的多組份納米復合物被細胞內化后,在經過白光(500mw/cm2)照射后,源自于dcf的綠色熒光顯著增強,說明光照后納米復合物中的微量cpts能夠顯著提升細胞內的ros水平,這對增強納米復合物的溶酶體逃逸具有重要的應用價值。
性能檢測4微量陽離子線性/支化聚噻吩構建的多組份納米復合物被細胞攝取后引起的溶酶體通透性變化。
利用質粒標記試劑盒(miruslabelit),對表達luciferase的質粒pluci進行rhodamine標記,得到rho-pluci。配制含微量cpt和rho-pluci的多組分納米復合物。實驗中,收集對數生長期的細胞,以7×104個/800μl的濃度接種于30mm玻璃培養皿中,置于含有5%co2的37℃培養箱內生長12-24h。當細胞融合70-80%時,將培養基吸除,pbs洗細胞1-2遍,加入含有載體材料/質粒的納米復合物(質粒終濃度為2μg/ml,n/p5)的不完全培養基。繼續孵育4h后,培養皿置于白光(500mw/cm2)下照射1min,對照組細胞則不經受光照。隨后使用lysotracker對細胞進行溶酶體染色。激光共聚焦顯微鏡觀察溶酶體熒光探針染料所用的激發波長為488nm,收集了505-525nm波長范圍的發射光;觀察羅丹明染料所用的激發波長為543nm,收集了560-700nm波長范圍的發射光。
結果如圖17a、17b、17c、17d、17e、17f至19a、19b、19c、19d、19e、19f所示,對照組pei/dna和sp/dna,在光照或非光照條件下,在細胞內均呈現很強的lysotracker綠色熒光,rho-pluci的紅色熒光與lysotracker綠色熒光高度重合,表明溶酶體膜比較完整,納米顆粒很難從溶酶體逃逸到胞漿。而攝取了微量cpt/sp/dna多組分納米復合物的細胞,在非光照時,lysotracker綠色熒光明顯變暗,表明溶酶體膜完整性受到擾動而導致ph值下降受阻;在光照后,lysotracker的綠色熒光非常暗淡,而且與rho-pluci的紅色熒光重合不佳,表明溶酶體膜完整性被破壞、質粒成功從溶酶體逃逸。該結果充分說明利用微量共軛聚合物的pci效應,能夠有效輔助納米復合物的溶酶體逃逸。
性能檢測5微量陽離子線性/支化聚噻吩構建的多組份納米復合物的invitro基因輸送實驗
收集對數生長期的細胞,調整細胞懸液濃度,以每孔大約1.6×104個/200μl的細胞濃度接種到96孔板,置于含有5%co2的37℃培養箱內生長12-24h。當細胞融合70-80%時,將培養基吸除,pbs洗細胞1-2遍,加入含有載體材料/質粒的納米復合物的不完全培養基,繼續孵育4-6h。隨后,吸除含有樣品的不完全培養基,pbs洗細胞1-2遍,每孔加入200μl完全培養基,培養箱內繼續孵育36-40h。對質粒pgfp的轉染效率通過熒光倒置顯微鏡觀察記錄(ex=465/95nm,em=515-555nm),對質粒pluc的轉染效率通過luciferaseassay(購買于美國promega公司)測定。
當檢測光照作用時,則在多組份納米復合物體系與細胞共孵育4~6h時引入光照條件即可,其余操作與未光照組一致。
陽離子線性/支化聚噻吩構建的多組份納米復合物體系對hela細胞的pgfp轉染40h。結果如圖20所示,當轉染過程中引入光照條件后,含有微量cpts的基因輸送體系對hela細胞的轉染效率顯著增強。結果表明,相比對照載體材料sp和pei,本發明使用含量為2.5%的cpt、結合pci策略可使luciferase的表達最多增強約10倍以上。
實施例3陽離子聚苯撐乙炔及其制備
本發明的陽離子聚苯撐乙炔的結構式為:
其中,cppe-det合成路線如下所示:
具體的制備方法如下:
(1)2,5-二碘-1,4-二苯乙酸乙酯的合成:將2,5-二碘-1,4-二苯乙酸(4.45g,10mmol)溶于無水乙醇(100ml),滴加二氯亞砜(1ml),室溫下攪拌24h,旋蒸除去溶劑,硅膠柱分離后得到淺黃色固體(4.0g);
(2)ppe-et合成:將2,5-二碘-1,4-二苯乙酸乙酯(0.5g,1mmol)溶于無水四氫呋喃(15ml)和三乙胺(5ml),脫氣后加入催化劑量的pd(pph3)4和cui,鼓氮氣10min,加入三甲基硅烷乙炔(0.27ml,2mmol),室溫下反應0.5h,向反應液滴加1m四丁基氟化銨溶液(2ml,2mmol),在室溫下繼續反應24h,將反應混合物通過短的硅膠柱,用乙醚沉淀3次,得到淺黃色固體(0.16g);
(3)cppe-det的合成:取78mg(0.2mmol)ppe-et溶于10ml干燥的nmp,冷卻后,加入二乙基三胺的nmp溶液(100eq,10ml),35℃反應3d,鹽酸中和后,先后用鹽酸(ph2)和水透析,最后冷凍干燥,得到黃色固體cppe-det。
在制備陽離子聚苯撐乙炔時,步驟(1)中,將2,5-二碘-1,4-二苯乙酸與二氯亞砜的摩爾比替換成10:1、50:1、100:1,均可制得該結構的陽離子聚苯撐乙炔;步驟(2)中,將2,5-二碘-1,4-二苯乙酸乙酯、pd(pph3)4、cui及三甲基硅烷乙炔的摩爾比替換為0.25:0.001:0.001:1、0.5:0.01:0.01:1,均可制得該結構的陽離子聚苯撐乙炔。
實施例4陽離子聚苯撐乙炔構建的多組份納米復合物
原料組成:該復合物包括聚氨基酸衍生物、rna及陽離子聚苯撐乙炔;其中,陽離子聚苯撐乙炔及聚氨基酸衍生物的正電荷總和與rna的負電荷之比為10:1,共軛聚電解質與聚氨基酸衍生物的正電荷之比為0.1%、0.25%、0.5%或1.0%。
其中,所述聚氨基酸衍生物,為聚賴氨酸或多聚精氨酸,分子量為5~50kda。
制備方法:在聚合物材料所含的胺基(包括1~4級胺)數目與核酸分子所含磷酸酯鍵數目之比為5的條件下(即n/p=5),首先將聚氨基酸和rna用超純水稀釋到相同的體積后,將rna溶液緩慢加入到聚氨基酸中,完全加完后靜置30min,使材料與質粒自組裝形成穩定的納米復合物;隨后,向納米復合物體系中緩慢加入等體積的陽離子聚苯撐乙炔,完全加完后再靜置60min,得到穩定的多組份納米復合物。
性能檢測6微量陽離子聚苯撐乙炔構建的多組份納米復合物的invitro基因輸送實驗
收集對數生長期的細胞,調整細胞懸液濃度,以每孔大約1.6×104個/200μl的細胞濃度接種到96孔板,置于含有5%co2的37℃培養箱內生長12~24h;當細胞融合70-80%時,將培養基吸除,pbs洗細胞1~2遍,加入含有載體材料/質粒的納米復合物的不完全培養基,繼續孵育4~6h;隨后,吸除含有樣品的不完全培養基,pbs洗細胞1-2遍,每孔加入200μl完全培養基,培養箱內繼續孵育36-40h,對pgfp的轉染水平通過熒光倒置顯微鏡觀察記錄(ex=465/95nm,em=515-555nm),對pluci的轉染效率通過luciferaseassaykit(promega公司)定量測定。
注意當檢測光照作用時,則在多組份納米復合物體系與細胞共孵育4-6h時引入光照條件即可,其余操作與未光照組一致。
微量陽離子聚苯撐乙炔的多組份納米復合物體系對hela細胞轉染40h的結果如圖21a、21b至26a、26b及圖27所示,cppe修飾后能夠顯著提高sp/dna復合物對pgfp和pluc轉染效率。0.1%、0.25%、0.5%、1.0%不同的cppe含量條件下轉染效率均有提升,0.25%的作用效果最佳。luciferaseassay檢測結果顯示,在不引入光照的條件下,pim-2就具有很強的促進sp/dna復合物轉染的能力,0.25%的pim-2材料加入后,多組份納米復合物的基因轉染效率相比sp提高了10倍,相比bpei提高了約23倍。上述結果均證實了微量陽離子聚苯撐乙炔構建的多組份納米復合物體系能夠顯著提高基因輸送效率。
實施例5陽離子聚苯撐乙烯及其制備
本發明的陽離子聚苯撐乙烯的結構式為:
其中,n=10~200。
其制備路線為:
制備方法包括如下步驟:
(1)2,5-二碘-1,4-二苯乙酸乙酯的合成:將2,5-二碘-1,4-二苯乙酸(4.45g,10mmol)溶于無水乙醇(100ml),滴加二氯亞砜(1ml),室溫下攪拌24h,旋蒸除去溶劑,硅膠柱分離后得到淺黃色固體(4.0g);
(2)ppv-et的合成:將2,5-二碘-1,4-二苯乙酸乙酯(0.5g,1mmol)、1,4-苯二乙烯(0.13g)溶于無水二氧六環(10ml),鼓氮氣30min;氮氣氣氛下加入三乙胺(1ml)和催化量5%pd/c,加熱回流,反應24h,過濾后旋蒸除去溶劑,氯仿溶解,在乙醚中沉淀3次,得到黃色固體(0.3g);
(3)cppv-det的合成:取78mg(0.2mmol)ppv-et溶于10ml干燥的nmp,冷卻后,加入二乙基三胺的nmp溶液(100eq,10ml),35℃反應3d;鹽酸中和后,先后用鹽酸(ph2)和水透析,最后冷凍干燥,得到黃色固體cppv-det。
在制備陽離子聚苯撐乙烯時,步驟(1)中,將2,5-二碘-1,4-二苯乙酸與二氯亞砜的摩爾比1替換成10:1、50:1、100:1,均可制得該結構的陽離子聚苯撐乙烯;步驟(2)中,將2,5-二碘-1,4-二苯乙酸乙酯與1,4-苯二乙烯的摩爾比替換為0.8:1、1:1、1.25:1,均可制得該結構的陽離子聚苯撐乙烯。
實施例6陽離子聚苯撐乙烯構建的多組份納米復合物
原料組成:該復合物包括聚酰胺衍生物、dna及陽離子聚苯撐乙烯;其中,陽離子聚苯撐乙烯及聚酰胺衍生物的正電荷總和與dna的負電荷之比為1:1,陽離子聚苯撐乙烯與聚酰胺衍生物的正電荷之比為0.01%。其中,所述聚酰胺衍生物為胺型聚酰胺pamam(第3.0~6.0代)。
制備方法:在聚合物材料所含的胺基(包括1~4級胺)數目與核酸分子所含磷酸酯鍵數目之比為5的條件下(即n/p=5),首先將聚酰胺衍生物和dna用超純水稀釋到相同的體積后,將質粒溶液緩慢加入到聚酰胺衍生物中,完全加完后靜置30min,使材料與質粒自組裝形成穩定的納米復合物。隨后,向納米復合物體系中緩慢加入等體積的陽離子聚苯撐乙烯,完全加完后再靜置30min,得到穩定的多組份納米復合物。
實施例7陽離子共聚物聚(芴-co-苯)及其制備
本發明的陽離子共聚物聚(芴-co-苯)的結構式為:
其中,n=10~200。
其制備路線為:
具體的制備方法包括如下步驟:
將fl-et(496mg,1mmol)、1,4-二苯硼酸(166mg,1mmol)溶于甲苯(10ml)和碳酸鉀水溶液(5ml,2m),脫氣后加入催化量的四(三苯基膦)鈀,90℃反應24h,氯仿萃取、水洗、無水硫酸鈉干燥,旋蒸濃縮,再經3次乙醚沉淀、干燥,得到淺黃色固體pfp-et(220mg),取82mg(0.2mmol)pfp-et溶于10ml干燥的nmp,冷卻后,加入r-nh2的nmp溶液(100eq,10ml),35℃反應3d,鹽酸中和后,先后用鹽酸(ph2)和水透析,最后冷凍干燥,得到黃色固體cpfp-r。
在制備陽離子共聚物聚(芴-co-苯)時,將fl-et、1,4-二苯硼酸與四(三苯基膦)鈀的摩爾比為替換為10:10:1,25:25:1,均可制得該結構的陽離子共聚物聚(芴-co-苯)。
實施例8陽離子共聚物聚(芴-co-苯)構建的多組份納米復合物
原料組成:該復合物包括聚糖衍生物、rna及陽離子聚(芴-co-苯);其中,陽離子聚芴及聚糖衍生物的正電荷總和與rna的負電荷之比為20:1,陽離子聚(芴-co-苯)與聚糖衍生物的正電荷之比為10%。其中,所述聚糖衍生物為殼聚糖。
制備方法:在聚合物材料所含的胺基數目與核酸分子所含磷酸酯鍵數目之比為5的條件下(即n/p=5),首先將聚糖和rna用超純水稀釋到相同的體積后,將質粒溶液緩慢加入到聚糖中,完全加完后靜置20min,使材料與質粒自組裝形成穩定的納米復合物,隨后,向納米復合物體系中緩慢加入等體積的陽離子聚(芴-co-苯),再靜置10min,得到穩定的多組份納米復合物。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 一種TaC@洋蔥狀碳/無定形碳納米復合物及其制備方法和應用與制造工藝
- 一種W2C@洋蔥狀碳/無定形碳納米復合物及其制備方法和應用與制造工藝
- 五氧化二礬及其碳納米復合物規模化制備與鋰電池應用的制造方法與工藝
- 一種ZnPc?UCNP?PEG?G納米復合物的制備方法與制造工藝
- 蒙脫土增強固體丙烯酸酯樹脂及其制備方法與制造工藝
- 一種Fe(Ⅲ)和Cu(Ⅱ)摻雜的聚苯胺納米復合物及其應用
- 一種Sn@C@g?C<sub>3</sub>N<sub>4</sub>納米復合物及其制備方法
- 一種Co@C@g?C<sub>3</sub>N<sub>4</sub>納米復合物及其制備方法和應用
- 基于超支化納米復合物的雙酚a增敏型免疫層析金標試紙條的制備方法
- 一種的強韌性石墨烯納米改性橡膠及其制備工藝的制作方法
- 一種基于生物功能化納米銀裝載紫杉醇或其類似物的靶向遞送系統的制作方法與工藝
- 一種Al?Zn?Si納米復合物速凝早強劑及其制備方法與流程
- 一種載納米銀的PNIPAM/PVA復合溫敏凝膠的制備及其應用的制作方法與工藝
- 一種利用藍莓葉提取液生物合成的納米銀抑菌劑及其制備工藝的制作方法與工藝
- 金屬?有機配合物與銀納米線復合物及其制備方法與應用與流程
- 一種TiO2/SnO2/Carbon納米復合物及其制備方法與流程
- 一種含納米NaY分子篩的復合物及其制備方法與流程
- 基于CdTe?Ag2Se納米復合物的皮質酮電化學發光傳感器的制備方法和應用與流程
- 一種以納米銀茶葉渣生物碳為主濾料的車載式空氣凈化器的制作方法與工藝
- PLGA納米復合物及其制備方法與流程