一種電活性硫雜多環芳烴化合物及其制備方法和應用與流程
本發明屬于富π共軛化合物領域,特別涉及一種電活性硫雜多環芳烴化合物及其制備方法和應用。
背景技術:
在材料學、化學和物理學中,其最重要的目標之一就是制備一些具有規整形狀的具有大π共軛體系的化合物。電活性材料(electroactivematerial,em)是電子-離子的混合導體,其體相在氧化和還原過程中能夠從溶液中可逆地置入和釋放離子(離子交換或充放電過程)成為電導體。em主要包括無機過渡金屬絡合物、導電高分子聚合物和有機/無機雜化材料,它們所具有的電化學屬性并不隨em的體相不同而改變。em在離子選擇性電極、離子交換膜、化學/生物傳感器、電催化、二次電池以及電致生色器件等新技術領域有著廣闊的應用前景。因此,尋求廉價、易合成、高電子傳遞速率、高離子交換容量、高電催化活性和高循環穩定性的em,是em研究的重點。開發擁有與常規硅等無機半導體材料不同特性的光活性材料的研究正飛速發展,尤其是利用π-電子共軛體系的有機單分子、高分子為基本組成單元的電活性材料,其設計和開發越來越活躍。與此相關聯,擁有規整結構、大的π-共軛體系的化合物及其衍生物正受到巨大的矚目。這一類富π共軛化合物根據其極性官能團的位置和取向,不僅能通過本身所擁有的極強的π-π相互堆積作用很容易發生自組裝,從而使大環形成固態一維柱狀非共價鍵高分子聚集體結構,固態二維網狀結構和三維結構。這種高度有序的一維、二維、三維結構可為空穴、電子等載體提供可能的移動通道,而且能夠被單電子氧化劑氧化而顯示具有光電活性。因此這類富π共軛化合物作為嶄新的顯示各向異性特性的電子及光電子材料素材而引起了科學家們的極大的興趣。
含s雜原子的多環芳烴化合物由于其潛在的功能和應用受到廣泛關注,它們具有非常規整的形狀結構,這類共軛化合物不僅能夠通過自身π-π堆砌或者s-s相互作用自組裝構筑一維、二維或三維超分子結構,而且能夠被單電子氧化劑氧化而顯示具有光電活性。
技術實現要素:
本發明所要解決的技術問題是提供一種電活性硫雜多環芳烴化合物及其制備方法和應用,該化合物作為優良的自組裝單元分子應用于有機發光二極管(oled)、有機場效應晶體管(ofet)、有機太陽能電池(osc)等高科技領域。
本發明的一種電活性硫雜多環芳烴化合物,所述化合物的結構通式為:
其中,r1、r2和r3各自獨立的選自疏水性長鏈基團、親水性長鏈基團、手性長鏈基團、酯基、氰基、氨基、巰基、羧基中的一種。
優選的,所述化合物為
最優選的,所述化合物為
本發明還提供了一種電活性硫雜多環芳烴化合物的制備方法,包括:
以3,5-二溴芐基溴為起始原料,通過和對甲基苯磺酰甲基異腈反應得到1,3-二(3,5-二溴芐基)丙酮,再與乙二酮類化合物發生羥醛縮合反應得到環戊二烯酮類化合物,再與對烷基二苯乙炔類化合物通過d-a反應得到2,3,4,5-四烷基苯基-1,4-二(3,5-二溴苯基)苯類化合物,再和噻吩硼酸酯衍生物進行suzuki偶聯反應,得到氧化前驅體,最后利用所得前驅體通過三氯化鐵作為氧化劑的scholl反應,制備得到電活性硫雜多環芳烴化合物i;
或者以2-溴苯乙酮為起始原料,在三氟甲磺酸的作用下發生羥醛縮合三聚反應合成1,3,5-三(2-溴苯基)苯,再和噻吩硼酸酯衍生物發生suzuki反應,得到氧化前驅體,最后利用所得前驅體通過三氯化鐵作為氧化劑的scholl反應,制備得到電活性硫雜多環芳烴化合物ii。
本發明還提供了一種電活性硫雜多環芳烴化合物的應用,應用于自組裝構筑一維、二維或三維超分子結構。
所述超分子結構應用于有機發光二極管oled、有機場效應晶體管ofet或有機太陽能電池osc。
本發明的化合物具有規整的分子結構,可通過自身π-π堆砌或者s-s相互作用自組裝構筑一維、二維或三維超分子結構。具有電子活性,能夠被單電子氧化劑氧化。
有益效果
本發明制備的電活性硫雜多環芳烴化合物,通過在骨架中引入硫雜原子,能夠通過改變分子的電子親合勢、偶極等在諸如晶體生長、液晶相及光電性能等方面可以呈現出獨特的理化性質,成為一類非常有潛力的新型有機功能分子;并且可作為優良的自組裝單元分子應用于有機發光二極管(oled)、有機場效應晶體管(ofet)、有機太陽能電池(osc)等高科技領域;具備原料合成方法簡單,制備工藝成熟,產率較高等優點,具有良好的應用前景。
附圖說明
圖1為實施例5產物1的maldi-tofmass圖;
圖2為實施例10產物2的maldi-tofmass圖;
圖3(a)為實施例5產物1的低分辨透射電子顯微鏡圖(甲苯,1mg/ml);(b)為產物1的高分辨透射電子顯微鏡圖(甲苯,1mg/ml);(c)為產物1的低分辨透射電子顯微鏡圖(四氫呋喃,1mg/ml);(d)為產物1的高分辨透射電子顯微鏡圖(四氫呋喃,1mg/ml);
圖4(a)為實施例10產物2的低分辨透射電子顯微鏡圖(四氫呋喃,1mg/ml);(b)為產物2的高分辨透射電子顯微鏡圖(四氫呋喃,1mg/ml);
圖5為實施例5產物1在液固界面的stm圖;
圖6為實施例10產物2在液固界面的stm圖;
圖7為實施例5產物1的循環伏安圖;
圖8為實施例10產物2的循環伏安圖;
圖9為實施例5產物1的dft理論計算圖;
圖10為實施例10產物2的dft理論計算圖;
圖11為實施例5不同當量c18h13br3cl6nsb下,產物1在二氯甲烷中(1.0×10-5m)的紫外光譜;
圖12為實施例10不同當量c18h13br3cl6nsb下,產物2在二氯甲烷中(1.0×10-5m)的紫外光譜。
具體實施方式
下面結合具體實施例,進一步闡述本發明。應理解,這些實施例僅用于說明本發明而不用于限制本發明的范圍。此外應理解,在閱讀了本發明講授的內容之后,本領域技術人員可以對本發明作各種改動或修改,這些等價形式同樣落于本申請所附權利要求書所限定的范圍。
實施例1
化合物4的合成
在氬氣保護下,往裝有磁力攪拌子的干燥兩口瓶中加入化合物3(1.414g,7.24mmol)和nah(60%,608mg,2.18mmol,2.1eq.)的混合物,加入新蒸的乙醚(10ml),反應10分鐘后,將3,5-二溴芐基溴(5g,2.1eq.)的二甲亞砜(10ml)溶液用干燥的注射器加入到反應體系中,20℃下反應一個小時,通過mass追蹤,無原料,加水淬滅反應,用ch2cl2萃取(50ml×3),合并有機相,蒸餾水洗(50ml×3),無水硫酸鎂干燥有機相,過濾除去硫酸鎂,旋蒸蒸除溶劑,轉入100ml兩口瓶中,氬氣保護,加入乙醚(15ml)和二氯甲烷(5ml)的混合溶劑,加入38%的鹽酸(10ml),20℃反應5分鐘,停止反應,用ch2cl2萃取(50ml×3),合并有機相,蒸餾水洗(50ml×3),飽和的食鹽水洗,無水硫酸鎂干燥有機相,過濾除去硫酸鎂,旋蒸蒸除溶劑,chcl3/ch3oh再沉淀,濾液旋干用硅膠層析柱提純(洗脫劑:pe/ch2cl2=3/1),合并共得白色固體2.5g,產率65%。
1hnmrδ7.62(s,2h),7.29(s,4h),3.72(s,4h).
實施例2
化合物6的合成
氬氣保護下,往裝有磁力攪拌子的兩口瓶中加入化合物4(140mg,0.27mmol)和化合物5(146mg,0.27mmol),加入新蒸的1,4-二氧六環(3ml),置于125℃下,待回流后,加入氫氧化四丁基季銨(1.0m,ch3oh溶液,0.27ml),反應10分鐘后,溶液變為酒紅色,tlc追蹤,無原料點,停止反應,待反應冷卻到室溫,用ch2cl2萃取(50ml×3),合并有機相,蒸餾水洗(50ml×3),無水硫酸鎂干燥有機相,過濾除去硫酸鎂,旋蒸蒸除溶劑,再經柱層析分離提純(洗脫劑:pe/ch2cl2=4/1),得紫紅色固體113mg,產率41%。
1hnmr(400mhz,cdcl3)δ7.51(t,j=1.7hz,1h),7.29(d,j=1.8hz,2h),7.26(s,1h),7.26(s,1h),7.03(d,j=8.2hz,2h),6.80(d,j=8.2hz,2h),2.59(t,j=7.5hz,2h),1.62–1.52(m,3h),1.25(s,17h),0.88(t,j=6.8hz,3h).
13cnmr(101mhz,cdcl3)δ197.96,156.75,144.77,134.13,132.89,131.55,129.05,128.90,128.42,122.57,122.40,35.73,31.94,31.04,29.71,29.67,29.65,29.53,29.38,29.02,22.71,14.13.
maldi-tofmass:calcd.forc53h64br4o[m]+:m/z=1032.1691;found:[m+na]+:1053.6042.
實施例3
化合物8的合成
將化合物7(264mg,0.51mmo)和化合物6(480mg,0.47mmol)置于50mlschlenk管中,抽真空充ar氣數次,加入2mlph2o,回流反應24小時,反應溶液由酒紅色變為棕黃色,mass追蹤,沒有原料且有目標產物生成,停止反應,待反應瓶冷卻到室溫,再經柱層析分離提純(洗脫劑:pe),得黃色固體500mg,產率71%。
1hnmr(400mhz,cdcl3)δ7.12(t,j=1.7hz,1h),6.86(d,j=1.7hz,2h),6.74(d,j=8.1hz,4h),6.67(d,j=8.1hz,4h),2.40(t,j=7.4hz,4h),1.44(dd,j=14.5,7.2hz,4h),1.33–1.13(m,40h),0.89(t,j=6.8hz,6h).
13cnmr(101mhz,cdcl3)δ144.28,140.39,140.27,138.41,136.71,133.19,130.96,130.59,127.11,120.86,35.40,31.96,31.23,29.77,29.75,29.70,29.56,29.41,28.89,22.72,14.14.
maldi-tofmass:calcd.forc90h122br4[m]+:m/z=1523.5820;found:[m+na]+:1546.2096,[m+k]+:1564.4425.
實施例4
化合物10的合成
在氬氣保護下,向裝有磁力攪拌子和回流裝置的50ml兩口瓶中依次加入化合物8(85mg,0.057mmol),化合物9(262mg,0.68mmol,12eq.),pd(pph3)4(22mg,0.017mmol,30%mol),加入甲苯(冷凍除氧)(30ml),2mol/lk2co3(冷凍除氧)(1ml),加熱至110℃回流,反應72小時后停止反應。冷卻至室溫,ch2cl2萃取(50ml×3),合并有機相,蒸餾水洗(50ml×3),無水硫酸鎂干燥有機相,過濾除去硫酸鎂,旋蒸蒸除溶劑,再經柱層析分離提純(洗脫劑:pe/ch2cl2=4/1),得黃色固體45mg,產率36%。
1hnmr(400mhz,cdcl3)δ7.20(s,1h),6.91(s,2h),6.81(d,j=7.8hz,4h),6.71(d,j=4.4hz,6h),6.63(s,2h),2.76(t,j=7.2hz,4h),2.33(t,j=7.0hz,4h),1.28(s,80h),0.90(t,j=6.3hz,12h).
13cnmr(101mhz,cdcl3)δ145.17,141.76,141.47,140.49,140.26,139.84,139.62,137.84,133.25,131.35,128.26,126.94,124.61,122.42,35.47,32.01,31.97,31.78,31.27,30.28,29.87,29.84,29.79,29.74,29.70,29.65,29.57,29.49,29.42,29.22,28.97,22.77,22.75,14.20.
maldi-tofmass:calcd.forc154h230s4[m]+:m/z=2207.6880;found:2208.7996.
實施例5
化合物1的合成
在氬氣保護下,往250ml兩口瓶中加入化合物10(80mg,0.036mmol),加入50ml干燥的二氯甲烷,氬氣鼓泡20分鐘,將無水氯化鐵(236mg,1.45mmol)溶解在2ml硝基甲烷中,注入到兩口瓶中,90分鐘后向反應液中加入甲醇淬滅反應,析出沉淀,氯仿溶解,過柱,先用乙酸乙酯作洗脫劑除去雜質,再用熱thf作洗脫劑,旋蒸溶劑后得到黑色固體,再用少量thf溶解,加入500ml甲醇,析出沉淀,過濾得到紅色固體33mg,產率42%。
maldi-tofmass:calcd.forc154h210s4[m]+:m/z=2188.5343;found:2188.6700.(圖1)
實施例6
化合物12的合成
在氬氣保護下,往25ml兩口瓶中加入2-溴苯乙酮(20g,100mmol)以及三氟甲磺酸(1.5g,10mmol),置于130℃下反應7小時后,停止反應。冷卻至室溫并加適量水,ch2cl2萃取,無水硫酸鎂干燥,再經柱層析分離提純(固定相:硅膠,洗脫劑:pe/ch2cl2=6/1),最后得黃色固體13g,產率72%。
1hnmr(400mhz,cdcl3)δ7.72(d,j=8.0hz,1h),7.54(s,1h),7.49(dd,j=7.6,1.5hz,1h),7.40(dd,j=10.8,4.2hz,1h),7.25(td,j=7.8,1.6hz,1h).
實施例7
化合物14的合成
在氬氣保護下,往100ml兩口瓶中加入化合物9(2g,5.29mmol),化合物13(1.65g,5.85mmol),pd(pph3)4(320mg,0.291mol),抽換氣三次后,加入冷凍除氧的甲苯(40ml)和碳酸鉀水溶液(2m,24ml),將反應體系置于110℃下回流24小時后,停止反應。ch2cl2萃取,無水硫酸鎂干燥,再經柱層析分離提純(固定相:硅膠,洗脫劑:pe),最后得白色固體1.85g,產率78%。
1hnmr(400mhz,cdcl3)δ7.49(d,j=8.5hz,2h),7.44(d,j=8.5hz,2h),7.13(d,j=3.5hz,1h),6.76(d,j=3.4hz,1h),2.83(t,j=7.6hz,2h),1.71(dt,j=15.1,7.5hz,2h),1.29(s,18h),0.91(t,j=6.6hz,3h).
13cnmr(101mhz,cdcl3)δ146.35,140.26,133.76,131.84,126.92,125.14,123.10,120.63,31.93,31.62,30.26,29.66,29.64,29.55,29.36,29.09,22.69,14.11.
實施例8
化合物15的合成
在氬氣保護下,往裝有磁力攪拌器的100ml兩口瓶中加入化合物14(300mg,0.74mmol),抽真空充ar氣數次,加入無水thf(30ml),-78℃下緩慢滴加n-buli(1.6m,thf溶液,0.50ml,0.81mmol),45分鐘后,加入b(ome)3(0.15ml,1.26mmol),-78℃下繼續反應2小時,取出反應瓶室溫攪拌過夜,加入hcl水溶液(2m,1ml),繼續反應1小時,真空蒸除thf,ch2cl2萃取,合并有機相,蒸餾水洗,無水mgso4干燥,真空下蒸除溶劑,得到無色液體240mg,直接投入下一步反應。
實施例9
化合物16的合成
在氬氣保護下,往50ml兩口瓶中加入化合物12(60g,0.056mmol),化合物15(140mg,0.37mmol),pd(pph3)4(4mg,0.021mol),抽換氣三次后,加入冷凍除氧的甲苯(40ml)和碳酸鉀水溶液(2m,1ml),將反應體系置于110℃下回流24小時后,停止反應,ch2cl2萃取,無水硫酸鎂干燥,再經柱層析分離提純(固定相:硅膠,洗脫劑:pe/ch2cl2=10/1),最后得到黃色固體50mg,產率69%。
1hnmr(400mhz,cdcl3)δ7.49(d,j=8.3hz,2h),7.35(q,j=7.6hz,2h),7.25–7.19(m,1h),7.12(d,j=3.5hz,1h),6.98(d,j=8.2hz,2h),6.94(d,j=7.5hz,1h),6.80(s,1h),6.75(d,j=3.5hz,1h),2.83(t,j=7.6hz,2h),1.78–1.64(m,2h),1.40–1.20(m,18h),0.90(t,j=6.8hz,3h).
13cnmr(101mhz,cdcl3)δ145.13,141.79,141.51,140.50,139.85,139.61,137.87,133.29,131.36,128.28,126.91,124.57,122.41,119.48,35.45,31.98,31.95,31.74,31.22,30.27,29.83,29.80,29.75,29.71,29.68,29.62,29.53,29.45,29.38,29.20,28.95,22.74,22.71,14.14.
maldi-tofmass:calcd.forc90h108s3[m]+:m/z=1284.7613;found:1284.9225.
實施例10
化合物2的合成
在氬氣保護下,往250ml兩口瓶中加入化合物16(80mg,0.062mmol),加入60ml干燥的二氯甲烷,一直通入氬氣鼓泡,將無水氯化鐵(363mg,2.23mmol)溶解在2ml硝基甲烷中,注入到兩口瓶中,120分鐘后向反應液中加入甲醇淬滅反應,析出沉淀,氯仿溶解,過柱,先用乙酸乙酯作洗脫劑除去雜質,再用熱thf作洗脫劑,旋蒸溶劑后得到黃色固體,再用少量thf溶解,加入500ml甲醇,析出沉淀,過濾得到黃色固體38mg,產率48%。
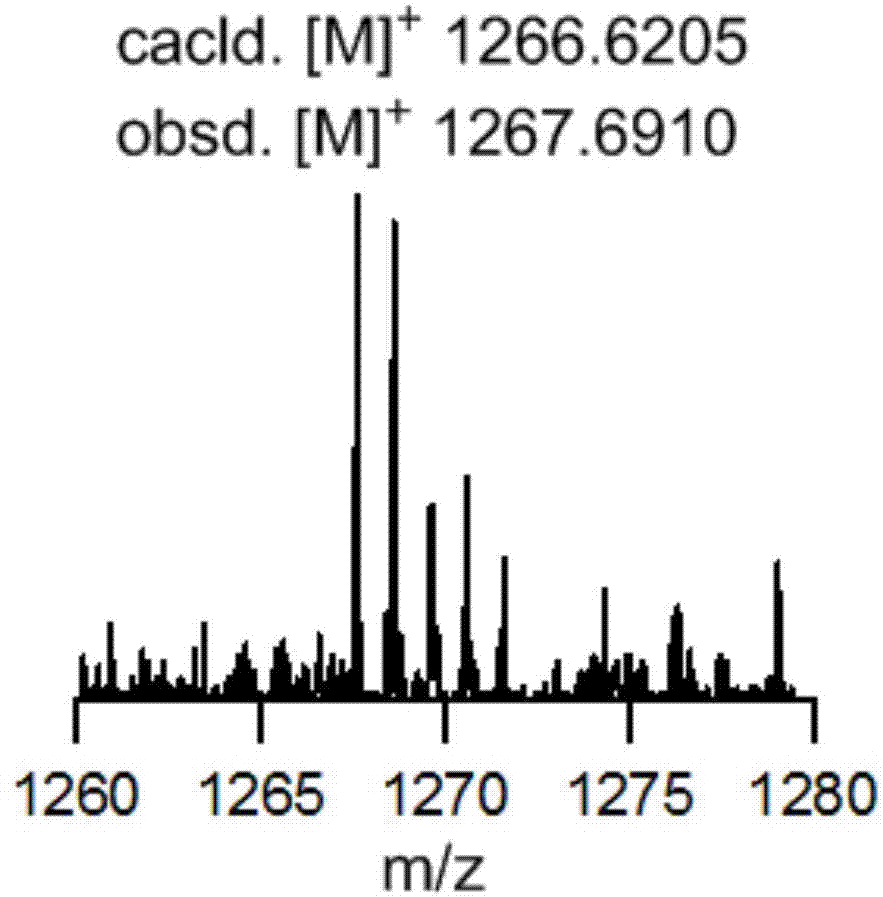
maldi-tofmass:calcd.forc90h90s3[m]+:m/z=1266.6205;found:[m+na]+:1267.6910.(圖2)
含s雜原子的多環芳烴化合物由于其潛在的功能和應用受到廣泛關注,它們具有非常規整的形狀結構,通過圖3-6可知,這類共軛化合物能夠通過自身π-π堆砌或者s-s相互作用自組裝構筑一維、二維或三維超分子結構。通過圖7-12可知,這類共軛化合物是很強的電子供體,所以在光伏器件中有著潛在的應用;并且這類共軛化合物具有較低的氧化電位,能夠被單電子氧化劑(c18h13br3cl6nsb)氧化而顯示具有光電活性。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 一種端巰基有機含磷?氮?硫酯化物及其制備方法和由其制得的阻燃聚酯纖維POY絲與流程
- 超小金屬硫族化合物納米晶、其親生物合成方法及應用與流程
- 一種端羧基有機含磷?氮?硫酯化物及其制備方法和由其制得的阻燃滌綸織物與流程
- 利用鈣磷化合物制備HA/β?TCP復合材料的方法與流程
- 含磷聚酯化合物組成及含磷阻燃環氧樹脂固化物的制備方法與流程
- 應用在環氧樹脂中含磷氮硫的反應型阻燃劑的制備方法與流程
- 高度有序結構的層狀金屬硫族化合物/碳納米管柔性復合薄膜材料及制備的制作方法與工藝
- 基于熱釋膠帶的二維過渡金屬硫屬化合物轉移方法與流程
- 一種含硫物檢測試紙的制作方法與工藝
- 對有機含磷化合物有毒氣體具有熒光響應的一維有機半導體納米線/帶及其制備方法和應用與流程