甲砜霉素四氟丙酸酯及其制備方法和應用與流程
本發明屬于醫藥及藥物分析技術領域,具體地說是涉及一種氟苯尼考中的未知雜質甲砜霉素四氟丙酸酯及其制備方法和應用。
背景技術:
氟苯尼考具有廣譜抗菌活性,可用于治療動物的革蘭氏陽性菌、革蘭氏陰性菌和立克次體等引起的感染,且對甲砜霉素、氯霉素的耐藥性細菌具有較強抑制作用,在體內殘留較低,現已被廣泛使用。其主要生產工藝流程如下:
即甲砜胺(i)與二氯乙腈進行成環縮合得到環合物(ii),用n,n-二乙基-1,1,2,3,3,3-六氟丙胺對環合物(ii)進行氟化得到氟化物(iii),氟化物(iii)水解得產物氟苯尼考。
按照《中華人民共和國獸藥典》(2010年版)采用hplc法分析氟苯尼考有關物質時,色譜記錄時間為主峰保留時間的2.5倍,而《中華人民共和國獸藥典》(2015年版)則將色譜記錄時間延長至主峰保留時間的5倍,保持其它條件與2010年版獸藥典相同。色譜記錄時間延長后,在相對保留時間rrt3.1處出現一較大未知雜質,按照2010年版獸藥典檢測則無法檢出該雜質。
氟苯尼考應用范圍廣泛,且用量大,因此對其所含雜質進行詳細研究并加以控制,將大大提高用藥安全性。對于采用2015年版中國獸藥典分析氟苯尼考新出現的相對保留時間rrt3.1未知雜質,各國獸藥典以及文獻、專利等均無該雜質的結構及制備等相關信息。
技術實現要素:
為了更好地控制氟苯尼考的質量,本發明通過對氟苯尼考中的未知雜質進行了分離、結構確證,提供了一種甲砜霉素四氟丙酸酯及其制備方法和應用。
本發明采用如下技術方案:
結構式如(iv)所示的甲砜霉素四氟丙酸酯:
上述結構式如(iv)所示的甲砜霉素四氟丙酸酯的結構通過下述方法判定得到:
取含有相對保留時間rrt3.1未知雜質的氟苯尼考樣品粉末與硅膠混合均勻,以石油醚-乙酸乙酯-氨水為淋洗劑,用硅膠層析柱過柱分離,收集含有目標未知雜質的洗脫液。
用2015版中國獸藥典中hplc法檢測上述洗脫液,其出峰時間與檢測氟苯尼考樣品時未知雜質的出峰時間吻合,說明收集的洗脫液所含物質為目標雜質。
經多次過柱富集,合并含目標雜質的洗脫液,減壓濃縮去除溶劑,剩余物料減壓干燥,得類白色固體。
將所得固體用甲醇溶解,利用esquire-lc型電噴霧離子阱質譜儀進行質譜分析。在正離子模式下通常可檢測到[m+h]+、[m+na]+、[m+k]+等離子峰,樣品在正離子模式下檢測到的m/z=483.9、506.2、522.1等離子峰符合該規律,說明該雜質的分子質量為483。
將樣品溶于dmso-d6,然后進行1h-nmr、13c-nmr、dept-135°、碳氫相關(hmqc)和氫氫相關(h-hcosy)分析。
在1h-nmr圖譜中,除了溶劑未氘代和水質子之外,共觀測到10組15質子信號,其中5質子化學位移在芳烴或不飽和質子區域,其余10質子的化學位移值在3~6.5ppm之間,指示該10質子附近均存在去屏蔽效應大(電負性大)的基團或原子。
在13c-nmr圖譜中觀測到13組碳信號(不計溶劑碳信號),其中芳烴區域有兩組強度大的碳信號。碳氫相關譜顯示芳烴區域兩組強度大的碳信號各與兩質子相關,同時dept-135°譜指示該兩碳不為2°碳,所以該兩組碳信號均由兩等價芳次甲基碳所貢獻,因此該雜質分子由15個碳組成。
結合氟苯尼考的制備工藝,根據雜質的esi+-ms、1h-nmr、13c-nmr、dept-135°圖譜、碳氫相關圖譜(hmqc)、氫氫相關圖譜(h-hcosy),推測該雜質為甲砜霉素四氟丙酸酯(iv),即2,3,3,3-四氟丙酸-[(2s,3r)-2-二氯乙酰氨基-3-羥基-3-(4-甲砜基苯基)]-丙酯,其分子式為c15h15cl2f4no5s,準確分子量為482.99,結構式如下:
本發明還提供了一種甲砜霉素四氟丙酸酯(iv)的制備方法,包括如下步驟:
(1)將(4r,5r)-2-二氯甲基-4,5-二氫-5-(4-甲砜基苯基)-4-噁唑甲醇(ii)與n,n-二乙基-1,1,2,3,3,3-六氟丙胺進行反應,得到化合物[(4r,5r)-2-二氯甲基-4,5-二氫-5-(4-甲砜基苯基)噁唑-4-基]甲醇-2,3,3,3-四氟丙酸酯,其結構式如(v)所示:
(2)[(4r,5r)-2-二氯甲基-4,5-二氫-5-(4-甲砜基苯基)噁唑-4-基]甲醇-2,3,3,3-四氟丙酸酯(v)水解開環得到甲砜霉素四氟丙酸酯(iv)。
作為優選,步驟(1)中,反應所用的溶劑為二氯甲烷、三氯甲烷、四氯化碳、1,2-二氯乙烷、四氫呋喃、甲基叔丁基醚、乙醚、甲苯、苯中的一種,反應溫度為-15~70℃,反應時間為3~24小時。
作為優選,步驟(1)中,反應所用的溶劑為二氯甲烷、三氯甲烷、四氫呋喃、甲基叔丁基醚、乙醚、甲苯中的一種,反應溫度為-5~40℃,反應時間為5~12小時。
作為優選,步驟(2)中,水解反應溶劑為水、乙醇-水、甲醇-水、異丙醇-水、正丁醇-水、乙腈-水、四氫呋喃-水中的一種,反應溫度為5~80℃,反應時間為1~8小時。
作為優選,步驟(2)中,水解反應溶劑為水、乙醇-水、異丙醇-水、乙腈-水、四氫呋喃-水中的一種,反應溫度為15~50℃,反應時間為2~5小時。
作為優選,甲砜霉素四氟丙酸酯的制備方法,具體包括如下步驟:
(1)反應瓶中投入16.9g(4r,5r)-2-二氯甲基-4,5-二氫-5-(4-甲砜基苯基)-4-噁唑甲醇及200g二氯甲烷,降溫至-5℃,然后緩慢滴加15gn,n-二乙基-1,1,2,3,3,3-六氟丙胺,滴加過程中控制物料溫度低于10℃,加完后于5~10℃攪拌反應8小時;反應結束,減壓蒸餾除去二氯甲烷,然后向剩余物中加入100ml冷水,攪拌,體系中原來的油狀物逐漸固化,過濾,并用冷水洗滌;所得濕品用50g甲醇溶解,降至-5~0℃攪拌結晶,過濾干燥,得[(4r,5r)-2-二氯甲基-4,5-二氫-5-(4-甲砜基苯基)噁唑-4-基]甲醇-2,3,3,3-四氟丙酸酯;
(2)反應瓶中加入上述制得的[(4r,5r)-2-二氯甲基-4,5-二氫-5-(4-甲砜基苯基)噁唑-4-基]甲醇-2,3,3,3-四氟丙酸酯15g,再加入100g水及30g乙醇,于40℃攪拌反應3小時;降溫至0℃結晶,過濾得濕品;所得濕品用80ml乙醇-水重結晶,真空干燥,得甲砜霉素四氟丙酸酯。
本發明用柱層析法對氟苯尼考中的未知雜質進行了分離,根據所分離雜質的ms-esi+、1h-nmr、13c-nmr、dept-135°圖譜、碳氫相關圖譜(hmqc)、氫氫相關圖譜(h-hcosy),判定該雜質為2,3,3,3-四氟丙酸-[(2s,3r)-2-二氯乙酰氨基-3-羥基-3-(4-甲砜基苯基)]-丙酯,即甲砜霉素四氟丙酸酯(iv)。
為了驗證結構的準確性,本發明根據氟苯尼考中的未知雜質的結構信息設計了合成路線,并成功制得了甲砜霉素四氟丙酸酯(iv)。產品經高效液相色譜法分析,其出峰時間與氟苯尼考樣品中未知雜質保留時間一致,經esi+-ms、1h-nmr分析,確證所得產物為氟苯尼考中的未知雜質甲砜霉素四氟丙酸酯(iv)。
本發明還提供了一種甲砜霉素四氟丙酸酯在氟苯尼考質量控制中的應用。在藥物研發和質量控制的過程當中,未知雜質的發現和制備往往對于該藥物品種的質量研究具有不容忽視的重要作用,可以將其應用于氟苯尼考生產工藝中的雜質定性及定量分析,從而可以提高并完善氟苯尼考的質量標準,可以大大提高其用藥安全性,為氟苯尼考的安全用藥提供重要指導意義。
本發明的有益效果在于:
本發明對氟苯尼考中未知雜質進行分離并通過esi+-ms、1h-nmr、13c-nmr、dept-135°圖譜、碳氫相關圖譜(hmqc)、氫氫相關圖譜(h-hcosy)等信息,確證了氟苯尼考中未知雜質為甲砜霉素四氟丙酸酯(iv),可以將該新雜質應用于檢測氟苯尼考原料藥或其制劑樣品的質量,完善氟苯尼考原料藥或其制劑的質量標準,可以大大提高用藥安全性。另外,本發明設計的甲砜霉素四氟丙酸酯(iv)合成工藝所用原料易得、反應條件溫和、反應過程容易控制、產品純度高。
附圖說明
圖1是本發明實施例2分離雜質的ms-esi+圖譜;
圖2是本發明實施例2分離雜質的1h-nmr圖譜;
圖3是本發明實施例2分離雜質的13c-nmr圖譜;
圖4是本發明實施例2分離雜質的dept-135°圖譜;
圖5是本發明實施例2分離雜質的hmqc圖譜;
圖6是本發明實施例2分離雜質的h-hcosy圖譜。
具體實施方式
下面結合附圖和具體實施例對本發明作進一步說明,但本發明的保護范圍并不限于此。
實施例1
取含有相對保留時間rrt3.1未知雜質0.83%的氟苯尼考樣品5g溶于甲醇,然后加入12g硅膠混合均勻,混合物于旋轉蒸發儀上減壓蒸出甲醇,得粉狀固體。將含有氟苯尼考樣品的粉狀固體均勻鋪于硅膠柱上端,以石油醚-乙酸乙酯-氨水(90:60:0.1)為淋洗劑進行過柱分離,用tlc監測不同餾分,收集含有相對保留時間rrt3.1雜質的餾分。
用2015版中國獸藥典中hplc法檢測該餾分,其主物質出峰時間與檢測氟苯尼考樣品時未知雜質的出峰時間吻合,說明此餾分所含物質為目標雜質。
重復上述操作,將得到的含有相對保留時間rrt3.1雜質的餾分合并,于50℃水浴中減壓濃縮去除溶劑,殘留物于45℃真空干燥,得類白色固體48mg。
實施例2
取少量實施例1所得固體用甲醇溶解,利用esquire-lc型電噴霧離子阱質譜儀進行質譜分析。樣品在正離子模式下檢測到的m/z=483.9、506.2、522.1等離子峰,分別為[m+h]+、[m+na]+、[m+k]+,因此該雜質的分子質量為483。
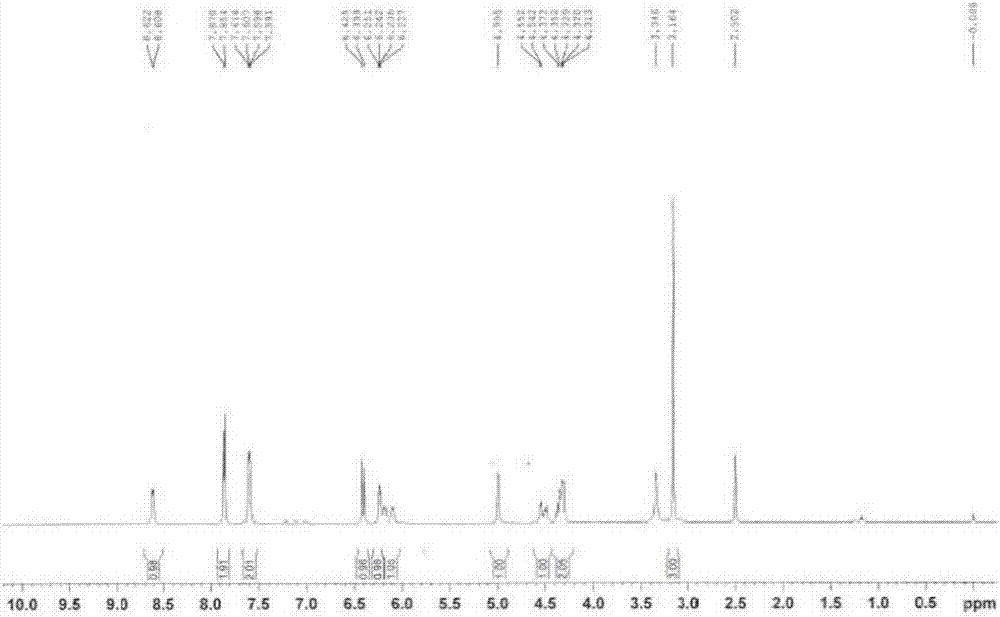
將樣品溶于dmso-d6,然后進行1h-nmr、13c-nmr、dept-135°、碳氫相關(hmqc)和氫氫相關(h-hcosy)分析,其結果如圖1~6所示。
在1h-nmr圖譜中,除了溶劑dmso未氘代和水質子之外,共觀測到10組15質子信號:1h-nmr(dmso-d6,500mhz)δh:8.62(d,j=7hz,1h),7.86(d,j=8hz,2h),7.60(dd,j=8.0,3.5hz,2h),6.41(d,j=13hz,1h),6.24(q,j=4.5hz,1h),6.09~6.19(m,1h),5.0(s,1h),4.35~4.55(m,2h),4.31~4.33(m,1h),3.16(s,3h)。上述質子有5質子化學位移在芳烴或不飽和質子區域,其余10質子的化學位移值在3~6.5ppm之間,指示該10質子附近均存在去屏蔽效應大(電負性大)的基團或原子。
在13c-nmr圖譜中觀測到13組碳信號(不計溶劑碳信號),13c-nmr(dmso-d6,125mhz)δc:44.1,53.7(d,j=8.9hz),66.1(d,j=35hz),66.7,70.2(d,j=13hz),84.1(m,j=hz),121.1(m,j=hz),127.0(強),127.6(強),140.1,148.2(d,j=4.8hz),162.3(d,j=22.8hz),164.2(d,j=3.3hz),其中127.0ppm及127.6ppm處的碳信號輕度大。碳氫相關譜(hmqc)顯示芳烴區域兩組強度大的碳信號各與兩質子相關,同時dept-135°圖譜指示該兩碳不為2°碳,所以該兩組碳信號均由兩等價芳次甲基碳所貢獻,因此該雜質分子由15個碳組成。
在dept-135°圖譜中,分子結構中的季碳不出峰,而仲碳則表現為倒峰,根據樣品的dept-135°圖譜信息,可以明確雜質分子中各碳原子的屬性。在氫氫相關譜(h-hcosy)中,可以確定質子化學位移和質子之間的偶合關系、連接順序。在碳氫相關譜(hmqc)圖譜中,可以明確各質子與連接碳的關系。從核磁共振譜中可以得到的信息如表1所示:
表1
結合氟苯尼考的制備工藝,根據雜質的esi+-ms、1h-nmr、13c-nmr、dept-135圖譜、碳氫相關圖譜(hmqc)、氫氫相關圖譜(h-hcosy),尤其是13c-nmr中δc121.1ppm、δc84.1pp處碳的裂分信息,推測該雜質為2,3,3,3-四氟丙酸-[(2s,3r)-2-二氯乙酰氨基-3-羥基-3-(4-甲砜基苯基)]-丙酯,即甲砜霉素四氟丙酸酯(iv),其分子式為c15h15cl2f4no5s,準確分子量為482.99,結構式為:
按照相關圖譜信息,各質子與碳信號的歸屬如表2所示:
表2
實施例3
制備甲砜霉素四氟丙酸酯(iv)工藝路線為:
(1)[(4r,5r)-2-二氯甲基-4,5-二氫-5-(4-甲砜基苯基)噁唑-4-基]甲醇-2,3,3,3-四氟丙酸酯(v)的制備
反應瓶中投入16.9g(4r,5r)-2-二氯甲基-4,5-二氫-5-(4-甲砜基苯基)-4-噁唑甲醇(ii)及200g二氯甲烷,降溫至-5℃,然后緩慢滴加15gn,n-二乙基-1,1,2,3,3,3-六氟丙胺,滴加過程中控制物料溫度低于10℃,加完后于5~10℃攪拌反應8小時。反應結束,減壓蒸餾除去二氯甲烷,然后向剩余物中加入100ml冷水,攪拌,體系中原來的油狀物逐漸固化,過濾,并用冷水洗滌。所得濕品用50g甲醇溶解,降至-5~0℃攪拌結晶,過濾干燥,得淡黃色固體18.5g,即[(4r,5r)-2-二氯甲基-4,5-二氫-5-(4-甲砜基苯基)噁唑-4-基]甲醇-2,3,3,3-四氟丙酸酯(v)。1h-nmr(dmso-d6,400mhz)δh:8.9(d,j=6.4hz,2h),7.64,(d,j=7.2hz,2h),7.28(s,1h),6.13~6.23(m,1h),5.78~5.84(m,1h)4,45~4,57(m,3h)3.23(s,3h)。13c-nmr(dmso-d6,100mhz)δc:43.9,67.1(d,j=7hz),73.0,82.5,83.9(dt,j=193hz,34hz),121.1(dd,j=280hz,26hz),126.8(強),128.1(強),141.5,145.4,162.2(d,j=25hz),162.6。ms(esi+):m/z465.9[m+h]+。
(2)甲砜霉素四氟丙酸酯(iv)的制備
反應瓶中加入上述過程制得的[(4r,5r)-2-二氯甲基-4,5-二氫-5-(4-甲砜基苯基)噁唑-4-基]甲醇-2,3,3,3-四氟丙酸酯(v)15g,再加入100g水及30g乙醇,于40℃攪拌反應3小時。降溫至0℃結晶2小時,過濾得濕品。所得濕品用80ml乙醇-水(4:1)重結晶,真空干燥,得類白色固體10.5g,即甲砜霉素四氟丙酸酯(iv)。1h-nmr(dmso-d6,400mhz)δh:8.61(d,j=7hz,1h),7.85(d,j=8.4hz,2h),7.59(dd,j=8.0,3.5hz,2h),6.40(d,j=13hz,1h),6.23(q,j=4.5hz,1h),6.08~6.19(m,1h),4.98(s,1h),4.34~4.54(m,2h),4.29~4.33(m,1h),3.15(s,3h)。ms(esi+):m/z483.9[m+h]+。
實施例4
一種甲砜霉素四氟丙酸酯的制備方法,具體包括如下步驟:
(1)[(4r,5r)-2-二氯甲基-4,5-二氫-5-(4-甲砜基苯基)噁唑-4-基]甲醇-2,3,3,3-四氟丙酸酯(v)的制備
反應瓶中投入16.9g(4r,5r)-2-二氯甲基-4,5-二氫-5-(4-甲砜基苯基)-4-噁唑甲醇(ii)及200g二氯甲烷,降溫至-5℃,然后緩慢滴加20gn,n-二乙基-1,1,2,3,3,3-六氟丙胺,滴加過程中控制物料溫度低于15℃,加完后于5~25℃攪拌反應8小時。反應結束,減壓蒸餾除去二氯甲烷,然后向剩余物中加入100ml冷水,攪拌,體系中原來的油狀物逐漸固化,過濾,并用冷水洗滌。所得濕品用50g甲醇溶解,降至-5~0℃攪拌結晶,過濾干燥,得淡黃色固體19.1g,[(4r,5r)-2-二氯甲基-4,5-二氫-5-(4-甲砜基苯基)噁唑-4-基]甲醇-2,3,3,3-四氟丙酸酯(v)。
(2)甲砜霉素四氟丙酸酯(iv)的制備
反應瓶中加入[(4r,5r)-2-二氯甲基-4,5-二氫-5-(4-甲砜基苯基)噁唑-4-基]甲醇-2,3,3,3-四氟丙酸酯(v)15g,再加入100g水及35g異丙醇,于45℃攪拌反應3小時。降溫至5℃結晶2小時,過濾得濕品。所得濕品用80ml異丙醇-水(4:1)重結晶,真空干燥,得類白色固體9.8g,即甲砜霉素四氟丙酸酯(iv)。
實施例5
甲砜霉素四氟丙酸酯在氟苯尼考質量控制中的應用
取氟苯尼考20mg,精密稱定,置10ml量瓶中,加流動相溶解并稀釋至刻度,搖勻,作供試品溶液;精密量取供試品溶液適量,加流動相稀釋制成每1ml中含氟苯尼考20μg的溶液作為對照溶液;精密稱取雜質甲砜霉素四氟丙酸酯適量,用流動相溶解并稀釋制成每1ml中含甲砜霉素四氟丙酸酯20μg的溶液作為對照品溶液。按照《中華人民共和國獸藥典》(2015年版)中,采用hplc法檢測。雜質甲砜霉素四氟丙酸酯和氟苯尼考的分離度應符合規定。精密量取對照品溶液10μl注入液相色譜儀,調節檢測靈敏度,使雜質峰的峰高為滿量程的10%~20%。再精密量取供試品溶液、對照溶液及對照品溶液各10μl,注入液相色譜儀,記錄色譜圖。
限度要求:供試品溶液的色譜圖中如有雜質峰,其中雜質甲砜霉素四氟丙酸酯(iv)的量按外標法以峰面積計算,不得超過0.15%;其余單個雜質峰(梯度溶劑峰不計)的面積不得超過對照溶液主峰面積的0.3倍(0.3%),總雜質不得過1.0%(供試品溶液中小于對照溶液主峰面積0.05倍的色譜峰可以忽略不計)。
藥物雜質檢查分析方法應靈敏、專屬,通過合適的分析技術將不同結構的雜質進行分離、檢測,從而達到對雜質的有效控制。本發明的優點是:操作簡便,測定結果準確可靠,專屬性強。
本方法同樣適用氟苯尼考制劑的有關物質分析。
- 還沒有人留言評論。精彩留言會獲得點贊!