一株可以高效分泌納豆激酶的枯草芽孢桿菌株及高純納豆激酶制備工藝的制作方法
本發明涉及一種微生物菌株及其發酵制備高純酶制劑的工藝,特別涉及一種高效分泌納豆激酶的枯草芽孢桿菌株及制備高純納豆激酶酶制劑的工藝。
背景技術:
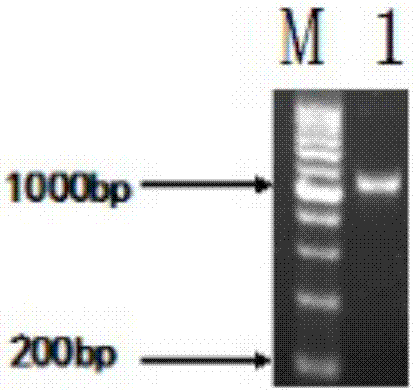
:納豆是黃豆經過納豆枯草桿芽孢桿菌發酵制成的發酵豆制品食品。納豆激酶(nk)是一種由納豆枯草芽孢桿菌產生的絲氨酸蛋白酶,存在于納豆中。nk除在體內外有強烈的纖維蛋白溶解活性外,還有促進血液流動、防血小板凝聚和降血壓等功效。nk于1980年首次由須見洋行博士發現,其相對分子質量(mr)為27000,等電點為8.6±0.3。由血栓引起的心血管疾病嚴重危害人類健康,目前溶栓劑包括注射類降解藥物(尿激酶、鏈激酶和組織型纖維蛋白原激活劑)和類組織纖維蛋白酶的蛋白(納豆激酶和蚓激酶)兩大類。注射類降解藥物高度依賴體內組織纖維蛋白酶原的水平,存在一定毒性,半衰期短且成本高。而nk屬于天然發酵產物,安全性高,具有體內作用時間長、價格低廉、有預防作用及易于規模化生產等優點,已發展為保健食品,其臨床應用已得到美國fda認可。提高nk工業化生產的產量和純度,對治療血栓疾病有重要意義。采用優化啟動子和信號肽、選用蛋白酶缺失的菌株以降低nk降解或者直接改造nk基因序列等技術手段以達到提高nk產量的目的。nk在枯草芽孢桿菌中可實現可溶性表達,wus等構建了枯草芽孢桿菌工程菌,并通過優化啟動子使nk的表達量增加了136%,達1999u/ml(平板法)。其他細菌,例如大腸桿菌、地衣芽孢桿菌、乳酸鏈球菌和酵母菌等也通過基因工程發酵nk。但這些技術也存在一些需要解決的問題,如在大腸桿菌中表達出沒有活性的包涵體;在其他工程菌中nk雖然能夠可溶性表達,但酶產量低,純化回收過程復雜,有待進一步改進工程菌構造。nishitoy等人已經證明,納豆發酵菌屬于枯草芽孢桿菌的一個亞種。枯草芽孢桿菌無致病性、無明顯的密碼子偏好性,具備包含轉錄、翻譯、蛋白質折疊和分泌等機制,常常作為工程菌株構建載體。本實驗前期驗證b.subtilis168含有nk基因序列,但不能表達有活性的nk。采用分子生物學方法,依據b.subtilis168的基因密碼子偏好性,優化nk-aprn基因編碼前30個氨基酸的序列,并加入六個組氨酸標記,以pht01為表達載體,成功構建工程菌,優化發酵條件后最高酶活可達289u/ml,高密度發酵發酵液中最高酶活達到7778u/ml,經親和柱層析制備高純納豆激酶酶制劑最高酶活為981731u/g,為納豆激酶基因工程進一步研究和納豆激酶的工業化生產奠定基礎。技術實現要素:本發明的目的在于克服天然菌種發酵性能的不足,提供一株可以高效分泌納豆激酶的枯草芽孢桿菌工程菌株和工程菌株用于發酵制備高純納豆激酶酶制劑的工藝。本研究利用pcr技術擴增納豆枯草芽孢桿菌的aprn基因,并依據枯草芽孢桿菌的密碼子偏愛性優化了起始30個氨基酸的密碼子,構建了重組表達質粒pht01-aprn,經限制性酶切、pcr擴增和測序驗證了其正確性。通過電擊法將pht01-aprn導入枯草芽孢桿菌168(bacillussubtilis168),利用氯霉素抗性篩選獲得工程菌。經iptg誘導表達,搖瓶發酵培養最高酶活為289u/ml,高密度發酵發酵液中最高酶活達到7778u/ml,經親和柱層析制備高純納豆激酶酶制劑最高酶活為981731u/g。為解決上述問題,本發明采用以下技術方案:一株可以高效分泌納豆激酶的枯草芽孢桿菌工程菌株,其特征在于,所述菌株的分類命名為:枯草芽孢桿菌(bacillussubtilis),保藏編號為cgmccno:13496,保藏單位為中國微生物菌種保藏管理委員會普通微生物中心(cgmcc),保藏地址為北京市朝陽區北辰西路1號院3號,保藏日期為2016年12月26日。本發明公開了構造高效分泌納豆激酶的枯草芽孢桿菌工程菌株的方法、搖瓶培養發酵產酶條件以及酶活測定方法,操作如下:步驟一:nk-aprn基因片段的擴增及優化以b.subtilis168菌液為第一次擴增的模版,以表1所列f1-r1引物對為第一次擴增引物,凝膠電泳驗證;膠回收產物為第二次pcr擴增模版,以表1所列f2-r1引物對為第二次擴增引物,凝膠電泳驗證;第二次膠回收產物為第三次pcr擴增模版,以表1所列f3-r2his引物對為第三次擴增引物,擴增出優化了前30個密碼子的aprn基因片段。步驟二:目的基因克隆步驟一擴增出的nk基因片段與pmd19-t載體連接,將連接產物采用熱擊方法轉化到e.colidh5α感受態中,涂布amp抗性平板。步驟三:重組質粒的構建及驗證質粒pht01和提取步驟二中陽性菌的克隆質粒雙酶切,用膠回收試劑盒回收純化目的片段,t4連接酶低溫過夜酶連,產物轉化e.colidh5α感受態細胞。菌落pcr鑒定后,取正確的陽性克隆活化培養,提取質粒測序。步驟四:工程菌的構建及鑒定制備b.subtilis168的感受態細胞,將步驟四中提取的質粒電轉感受態細胞。隨機挑取氯霉素抗性平板上長出的5-8個單菌落,菌落pcr鑒定,取正確的陽性克隆活化培養,提取質粒測序。步驟五:工程菌發酵活化工程菌培養種子液,以氯霉素作為抗生素,種子液轉接發酵培養基培養,od600在0.8-1.0降溫至25℃至30℃,加入終濃度為0.2-1.0mm的iptg誘導培養。步驟六:納豆激酶酶活的測定本發明采用四肽底物法檢測納豆激酶酶活。制作對硝基苯胺的標準曲線。nk活性單位被定義為nk每分鐘釋放1nmol硝基苯胺的含量。本發明還公開了工程菌高密度發酵的發酵產酶條件和高純納豆激酶酶制劑的制備工藝,操作如下:(1)高密度發酵接種:活化菌種,在37℃、200rpm培養條件下制備一級種子液和二級種子液,種齡為12h的二級種子液按照10%接種量接種至發酵培養基(氯霉素終濃度為5μg/ml、消泡劑0.01%)。發酵培養基(1l):葡萄糖28.5g,甘油57g,酵母提取物60g、吡啶二羧酸0.167g、nh4cl3g、k2hpo4·3h2o1g、mnso4·h2o0.032g、feso4·7h2o0.05g、cocl2·6h2o0.01g、zncl20.01g、mgso4·7h2o2g、cacl2·2h2o5g,補水至1000ml。發酵:攪拌速率初始設置為800rpm,通氣量為12l/min。設置參數,控制罐內溫度為37℃、溶氧量為30%、發酵液的ph7.0,依據溶氧情況調整攪拌速率和通氣量。蛋白誘導表達:取樣檢測od600,至對數前期降溫至28℃,加入iptg開始誘導發酵。補料:補料培養基(1l):甘油250g,酵母提取物100g,補水至1000ml。檢測發酵液od600和發酵培養基中甘油的消耗情況,分別采用一次補料和連續補料方式補料培養。終止發酵:發酵培養24-28h后,終止發酵。(2)高純納豆激酶酶制劑的制備發酵液經冷凍離心后收集上清液、(nh4)2so4沉淀去除雜蛋白并沉淀nk、鎳柱純化、脫鹽并濃縮、加冷凍保護劑和冷凍干燥等步驟,制成高純度納豆激酶酶制劑,納豆激酶酶活達到981713u/g。本發明具有如下的有益效果:構造枯草芽孢桿菌的納豆激酶工程菌,實現納豆激酶在其胞外表達,經優化發酵培養條件,酶活高達289u/ml,可進一步用于工業化液體發酵生產。附圖說明表1nk-aprn基因pcr擴增引物。表2nk-aprn基因pcr擴增體系。圖1瓊脂糖凝膠電泳鑒定nk-aprn基因pcr擴增電泳圖。圖2瓊脂糖凝膠電泳鑒定aprn基因克隆pcr擴增電泳圖。圖3瓊脂糖凝膠電泳鑒定pmd19-t:aprn質粒酶切電泳圖。圖4瓊脂糖凝膠電泳鑒定pht01質粒酶切電泳圖。圖5瓊脂糖凝膠電泳鑒定pht01:aprn質粒構建電泳圖。圖6瓊脂糖凝膠電泳鑒定工程菌構建電泳圖。圖7對硝基苯胺標準曲線。圖8工程菌酶活和細胞生長圖。圖9nk高密度發酵od600、酶活。具體實施方式以下對本發明的實施例作詳細說明:本實施例在以本發明技術方案為前提下進行實施,給出了詳細的實施方式和過程,但本發明的保護范圍不限于下述的實施例。下列實施例中未注明具體條件的實驗方法,通常按照常規條件,例如sambrook等分子克隆:實驗室手冊(newyork:coldspringharborlaboratorypress,1989)中所述的條件,或按照制造廠商所建議的條件。實施例1步驟一:nk-aprn基因片段的擴增及優化lb液體培養基:10g/l蛋白胨、10g/l氯化鈉、5g/l酵母提取物。lb固體培養基:lb液體培養基、15g/l瓊脂糖。amp溶液:100mg/ml。根據genbank收錄納豆激酶基因序列(gi:608796180),依據宿主菌b.subtitle密碼子偏好(http://www.kazusa.or.jp/codon/cgi-bin/showcodon.cgi?species=1423),利用生物軟件oligo設計引物(表1),擴增aprn基因并優化它的前30個氨基酸密碼子。引物由生工生物工程有限公司合成。將保藏的b.subtilis168菌株活化后,按照2%的接種量接種于lb培養基中,37℃、200rpm培養12h。目的基因的克隆采用over-lappingpcr的方法,即以菌液為第一次pcr(德國eppendrof公司)擴增模版,將凝膠電泳驗證后的膠回收(德國eppendrof公司)產物作為第二次pcr擴增模版,再以此次膠回收產物為模版進行第三次pcr擴增。引物依次為f1-r1、f2-r1和f3-r2his,擴增并優化源于b.subtilisnatto的aprn基因,如圖1。擴增引物如表1,組合引物依次為引物依次為f1-r1、f2-r1和f3-r2his,擴增體系如表2,擴增反應條件:94℃預變性10min,94℃變性40s,54℃退火40s,72℃延伸30s,共35個循環,72℃延伸10min。步驟二:目的基因克隆基因克隆方法參照《分子克隆實驗指南》。試劑盒回收并純化第三次pcr擴增產物,產物通過ta克隆連到質粒pmd19-t。pmd19-t載體連接體系:dna4.5μl,pmd19-t0.5μl,緩沖溶液i5μl,合計10μl,4℃過夜連接。通過熱激轉化方法將連接產物導入大腸桿菌感受態中,涂布amp抗性平板(終濃度100μg/ml)。提取陽性克隆菌落質粒dna,通過限制性內切酶酶切圖譜分析和pcr產物測序驗證,將正確的攜帶pmd19-t:aprn的質粒菌株置于甘油管-80℃保存。步驟三:重組質粒的構建及工程菌構造pmd19-t:aprn由限制性內切酶ecorv和bamhi酶切,切下的aprn基因片段插入pht01的smaⅰ和bamhi酶切位點之間,通過熱激轉化方法將連接產物導入大腸桿菌感受態中,涂布amp抗性平板(終濃度100μg/ml)。提取陽性克隆菌落質粒dna,通過限制性內切酶酶切圖譜分析和pcr產物測序驗證,將正確的攜帶pht01:aprn的質粒菌株置于甘油管-80℃保存。步驟四:工程菌的構建及鑒定b.subtilis168的感受態細胞制備和電擊轉化參考xue的方法。隨機挑取氯霉素抗性平板上長出的單菌落,提取陽性克隆菌落質粒dna,通過限制性內切酶酶切圖譜分析和pcr產物測序驗證,將正確的攜帶pht01:aprn的質粒菌株置于甘油管-80℃保存。步驟五:納豆激酶工程菌發酵:發酵培養基:20g/l葡萄糖、20g/l酵母提取物、2g/lk2hpo4·3h2o、1g/lmgso4·7h2o,2%甘油和1mm吡啶二羧酸,ph調至7.0-8.0。取甘油管中工程菌以2%接種量接種于lb培養基(含5μg/ml氯霉素)中,200rpm、37℃過夜培養。以2%接種量將種子液接種于25ml發酵培養基a中,37℃、200rpm搖床培養,至od600在0.8-1.0之間,降溫至25℃。接入終濃度為0.5mm的誘導劑iptg,在不同發酵時間測定菌液的od600和nk酶活,如圖8。步驟六:四肽底物法檢測酶活首先制作對硝基苯胺的標準曲線:配制對硝基苯胺樣品溶液,濃度分別為0.02、0.03、0.04、0.06和0.08mmol,在405nm下測定不同濃度硝基苯胺樣品溶液的吸光值,以水作為空白對照。發酵液4℃、8000r/min離心1min,將上清稀釋成不同濃度作為檢測樣品液。檢測方法在文獻基礎上加以改進:取100μl樣品稀釋液與50μl終濃度為0.5mm的四肽底物(d-val-leu-lys-對硝基苯胺)混合并在37℃水浴下反應1min,加入75μl50%的冰醋酸終止反應,在405nm下測定反應液吸光值。nk活性單位被定義為nk每分鐘釋放1nmol硝基苯胺的含量。實施例2接種:活化菌種,在37℃、200rpm培養條件下制備一級種子液和二級種子液,種齡為12h的二級種子液按照10%接種量接種至發酵培養基(氯霉素終濃度為5μg/ml、消泡劑0.01%)。發酵培養基(1l):葡萄糖28.5g,甘油57g,酵母提取物60g、吡啶二羧酸0.167g、nh4cl3g、k2hpo4·3h2o1g、mnso4·h2o0.032g、feso4·7h2o0.05g、cocl2·6h2o0.01g、zncl20.01g、mgso4·7h2o2g、cacl2·2h2o5g,補水至1000ml。發酵:攪拌速率初始設置為800rpm,通氣量為12l/min。設置參數,控制罐內溫度為37℃、溶氧量為30%、發酵液的ph7.0,依據溶氧情況調整攪拌速率和通氣量。蛋白誘導表達:檢測od600,當od600等于10時降溫至28℃加入終濃度為0.2-1.0mmiptg進行蛋白質誘導表達。補料:補料培養基(1l):甘油250g,酵母提取物100g,補水至1000ml。連續補料培養,設置補料程序為:發酵3h,od600=10開始補料,11-14h補料速率為250ml/h補料4h,15-18h再以170ml/h的速率補料4h,19-20h最后以170ml/h的速率補料2h。終止發酵:發酵培養24h后,終止發酵。經檢測,發酵20h酶活可達到7778.02iu/ml(rsd%=0.8),od600為173.5±0.85,見圖9。實施例3發酵液經4℃、9000r/m冷凍離心10min,取上清液;將研細的(nh4)2so4按照20%的飽和度加入發酵上清液中,4℃下保存過夜,10000r/m離心30min,棄去沉淀;繼續向發酵上清液中加硫酸銨至60%飽和度,4℃下保存過夜,10000r/m離心30min,棄去上清液,回收率達到72%。實施例4平衡鎳柱:吸取1ml含鎳填料至純化柱中,先用8倍體積去離子水清洗2遍,再用8倍體積結合液清洗三次。孵育:結合液將近流盡時,加入60ml發酵上清液,至4℃垂直旋轉冰箱孵育4h,帶有histag標簽的nk與填料結合。漂洗:下端排盡未與鎳柱結合的液體,加入10倍柱體積的漂洗液洗雜蛋白,在冰上垂直反轉5min,重復4次。洗脫:用移液槍吸取1.5ml洗脫液垂直攝入填料正中間將填料充分垂懸,冰上靜置2min,收集流出液。重復洗脫10次左右。洗脫液采用5kd的超濾管,300g離心,以純水作為稀釋液,以達到濃縮納豆激酶和除鹽的目的。向濃縮納豆激酶溶液中加入一定濃度冷凍保護劑,冷凍干燥即制得高純度納豆激酶酶制劑,經檢測納豆激酶酶活達到981713u/g。表1nk-aprn基因pcr擴增引物注:加下劃線和波浪線部分表擴增重復;黑體加粗并加下劃線部分表示酶切位點;斜體加下劃線部分表示加入六個組氨酸。表2nk-aprn基因pcr擴增體系組分體積(μl)基因組dna模板2引物-f1引物-r1dntp210×buffer2.5taqdna聚合酶0.25h2o16.25total25當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1