芘噻唑并噻唑基有機多孔聚合物及其制備方法與流程
本發明涉及有機材料技術領域,尤其涉及一種有機骨架材料及其制備方法。
背景技術:
多孔有機骨架材料具有相互連通的網狀多孔結構,因此相對于其他材料具有比表面積大和密度更小的優勢,被廣泛的應用于非均相催化、氫氣儲存、吸附、離子交換、氣體分離等領域。隨著多孔有機骨架材料越來越受到人們的關注,其優質的材料特性吸引更多的人去探究。
多孔有機骨架材料分金屬-有機骨架(mofs)和共價有機骨架(cofs)。金屬-有機骨架(mofs)又稱雜合化合物或配位聚合物,它是利用有機配體與金屬離子之間的金屬-配位絡合作用通過自組裝形成的具有超分子微孔網狀結構的材料。由于有機配體種類多樣,金屬離子選擇多樣,使得合成mofs材料具有很大的多樣性、靈活性。此外,還可以通過合成特殊的有機配體與特定的金屬離子裝配形成專一化的有機骨架。
然而,mofs材料的一些缺點導致其在應用上存在局限性:(1)在mofs失去溶劑分子之后其結構開始變得不穩定,含有多孔骨架的結構容易坍塌,導致孔徑縮小或者形成無定形的無孔結構;(2)對環境敏感:mofs材料中大多數對氣體中的水分子極其敏感,接觸久了可能會導致結構的變化設置骨架的坍塌;(3)結構表征比較困難;(4)可調節的孔徑范圍小:介孔區域的材料對有機配體結構長度要求較高,但配體長度的增加伴隨著溶解性質的變差。
由于mofs材料暴露出來的問題使得該方向的發展受到了阻礙,因此技術人員現在將研究重點放在了共價有機骨架(cofs)上。然而,現有技術中共價有機骨架材料的種類很少,大大限制了共價有機骨架的發展。
技術實現要素:
本發明的目的在于提供一種有機骨架材料及其制備方法,本發明提供的新的有機骨架材料拓展了有機骨架的范圍。
為了實現上述發明目的,本發明提供以下技術方案:
本發明提供了一種有機骨架材料的制備方法,包括如下步驟:
1,3,6,8-四(對甲酰基苯基)芘和二硫代乙酰胺在加熱條件下進行聚合反應,得到有機骨架材料;
所述有機骨架材料具有如下重復單元:
優選的,所述聚合反應的加熱溫度為120-200℃。
優選的,所述聚合反應的加熱溫度為140-180℃。
優選的,所述聚合反應在一種有機溶劑的存在下進行。
優選的,所述有機溶劑包含n,n-二甲基甲酰胺、硝基甲烷或苯酚的至少一種。
優選的,所述聚合反應在一種惰性氣氛下進行。
優選的,所述惰性氣氛為氮氣氣氛。
本發明還提供了一種上述技術方案所述制備方法得到的有機骨架材料,為含有芘環和噻唑并噻唑環的大分子網狀交聯聚合物。
附圖說明
圖1為本發明得到的1,3,6,8-四溴芘的紅外譜圖;
圖2為本發明得到的1,3,6,8-四(對甲酰基苯基)芘的紅外譜圖;
圖3為本發明得到的有機骨架材料的紅外譜圖;
圖4為有機骨架材料的熱重曲線;
圖5為有機骨架材料的10.0k的sem圖;
圖6為有機骨架材料的5.0k的sem圖。
具體實施方式
本發明提供了一種有機骨架材料的制備方法,包括如下步驟:
1,3,6,8-四(對甲酰基苯基)芘和二硫代乙酰胺在加熱條件下進行聚合反應,得到有機骨架材料;
所述有機骨架材料具有如下重復單元:
本發明對所述1,3,6,8-四(對甲酰基苯基)芘和二硫代乙酰胺的來源沒有特殊限制,采用商品試劑作為反應物,或者按照文獻上合成方法制備反應物均可。
在本發明中,1,3,6,8-四(對甲酰基苯基)芘和二硫代乙酰胺可以在一種有機溶劑的存在下反應。所述有機溶劑優選為n,n-二甲基甲酰胺、硝基甲烷或苯酚中的一種或幾種。在本發明中,當所述有溶劑包含兩種、三種或四種組分時,所包含的各物質可以以任意質量分數存在。
在本發明中,所述聚合反應的溫度優選為120~200℃,更優選為140~180℃。
在本發明中,所述聚合反應可以在一種惰性氣氛下進行。優選的,所述惰性氣氛為氮氣氣氛。
在本發明中,所述1,3,6,8-四(對甲酰基苯基)芘和二硫代乙酰胺的摩爾比優選為(1.5~2.5):1,更優選為(1.7~2.3):1,進一步優選為(1.9~2.1):1。
本發明優選對聚合產物進行后處理,得到有機骨架材料。在本發明中,所述后處理包含洗滌和干燥。
在本發明中,所述洗滌可以使用有機溶劑。所述有機溶劑可以是n,n-二甲基甲酰胺(dmf)和四氫呋喃(thf)中的一種或混合物,也可以采用本領域技術人員所熟知的其他有機溶劑。
本發明對所述干燥沒有特殊要求,具體的可以真空干燥、紅外燈下烘干、室溫晾干或者加熱爐干燥。本發明對所述干燥的溫度和時間沒有特殊要求,能夠使洗滌所用的有機溶劑揮發干凈即可。
本發明還提供了一種有機骨架材料,其特征在于,所述有機骨架材料具有如下結構單元:
下面結合實施例對本發明提供的有機骨架材料及其制備方法進行詳細的說明,但是不能把它們理解為對本發明保護范圍的限定。
在一些實施例中,可以使用有機溶劑。有機溶劑可以是n,n-二甲基甲酰胺(dmf)、硝基甲烷或者苯酚中的一種或多種。也可以不使用有機溶劑。在加熱條件下,反應物可以熔化而形成混合物進行聚合反應。
實施例1
用藥匙取芘(5.0369g,24mmol)置于潔凈的燒杯中,量取硝基苯(100ml)倒入燒杯中攪拌溶解,待完全溶解后開始以滴狀緩慢滴加液溴(5.6ml,109mmol),然后將反應體系升溫至120℃,反應時間為14小時,反應液顏色轉變成土黃色,過濾得到的粗產品用乙醇超聲清洗3次,得到淡綠色固體1,3,6,8-四溴代芘,產量為6.2687g。
實施例2
向圓底燒瓶(250ml)中加入氯化鈀(0.0942g),三苯基膦(0.643g)和二甲亞砜(7.5ml)溶液,在氮氣的保護下,將反應體系加熱至固體物質全部溶解為止,整個溶液呈現黃色,并且透明。反應溫度在130℃到140℃左右,關閉油浴,加入水合肼(4滴)后,有亮黃色固體物質析出,快速過濾,在分別用乙醇(5ml*2)和無水乙醇(5ml*2)進行洗滌,真空干燥后得到亮黃色固體0.2675g,氮氣下保存。
實施例3
混合1,3,6,8-四溴芘(1.0039g),4-對甲酰基苯基硼酸(1.7540g),鈀四催化劑(0.2675g)和碳酸鉀(2.2044g)在干燥的四氫呋喃(30ml)在氮氣下并在85℃下攪拌三天,然后將黃色懸浮液混合物傾入冰中并攪拌,冰中含有濃鹽酸,再將混合物進行過濾,過濾后的產物用2m鹽酸洗滌3次(20ml),洗過的產品繼續用氯仿(3*100ml)萃取,取下層溶液后用硫酸鎂干燥,過濾后,將溶劑在減壓下得到的固體殘留物,在氯仿中重結晶,得到一種亮黃色產物1,3,6,8-四(對甲酰基苯基)芘(0.2355g)。
實施例4
在聚四氟乙烯襯里的高壓釜中加入1,3,6,8-四(對甲酰基苯基)芘0.1230g(0.20mmol)和二硫代乙酰胺0.0480g(0.40mmol)。再加入3ml無水dmf,充氮氣5分鐘,密封好反應釜后。置于烘箱中,溫度控制在200℃反應12小時,冷卻至室溫過濾,用dmf洗滌2次除去未反應的物質,反應結束后,產物通過四氫呋喃索氏提取24小時,產物干燥保存。
實施例5
在聚四氟乙烯襯里的高壓釜中加入1,3,6,8-四(對甲酰基苯基)芘0.1230g(0.20mmol)和二硫代乙酰胺0.0480g(0.40mmol)。再加入3ml無水硝基甲烷與dmf體積比為1:1的混合溶劑,充氮氣5分鐘,密封好反應釜后。置于烘箱中,溫度控制在180℃反應12小時,冷卻至室溫過濾,用dmf洗滌2次除去未反應的物質,反應結束后,產物通過四氫呋喃索氏提取24小時,產物干燥保存。
實施例6
在聚四氟乙烯襯里的高壓釜中加入1,3,6,8-四(對甲酰基苯基)芘0.1230g(0.20mmol)和二硫代乙酰胺0.0600g(0.50mmol)。再加入3ml無水dmf,充氮氣5分鐘,密封好反應釜后。置于烘箱中,溫度控制在160℃反應24小時,冷卻至室溫過濾,用dmf洗滌2次除去未反應的物質,反應結束后,產物通過四氫呋喃索氏提取24小時,產物干燥保存。
實施例7
在聚四氟乙烯襯里的高壓釜中加入1,3,6,8-四(對甲酰基苯基)芘0.1230g(0.20mmol)和二硫代乙酰胺0.0480g(0.40mmol)。再加入3ml無水苯酚,充氮氣5分鐘,密封好反應釜后。置于烘箱中,溫度控制在140℃反應36小時,冷卻至室溫過濾,用dmf洗滌2次除去未反應的物質,反應結束后,產物通過四氫呋喃索氏提取24小時,產物干燥保存。
實施例8
在聚四氟乙烯襯里的高壓釜中加入1,3,6,8-四(對甲酰基苯基)芘0.1230g(0.20mmol)和二硫代乙酰胺0.0480g(0.40mmol)。再加入3ml無水硝基甲烷,充氮氣5分鐘,密封好反應釜后。置于烘箱中,溫度控制在120℃反應48小時,冷卻至室溫過濾,用無水硝基甲烷洗滌2次除去未反應的物質,反應結束后,產物通過四氫呋喃索氏提取24小時,產物干燥保存。
實施例9
在聚四氟乙烯襯里的高壓釜中加入1,3,6,8-四(對甲酰基苯基)芘0.1230g(0.20mmol)和二硫代乙酰胺0.0480g(0.40mmol)。充氮氣5分鐘,密封好反應釜后。置于烘箱中,溫度控制在120℃反應48小時,冷卻至室溫過濾,用無水硝基甲烷洗滌2次除去未反應的物質,反應結束后,產物通過四氫呋喃索氏提取24小時,產物干燥保存。
醛基與二硫代乙酰胺的聚合反應可能遵循以下歷程:
由反應歷程可知,如果反應前驅體中含有多個醛基,最終可能生成鏈狀或網狀聚合物。
1,3,6,8-四(對甲酰基苯基)芘具有四個基本位于一個平面上的醛基,因此,反應后的產物也是一個近平面的網狀聚合物。
性質與表征
通過圖1對1,3,6,8-四溴芘結構進行紅外圖譜分析,圖1中出現了主要的-c-br的特征峰(653.87cm-1),說明-br基出現在了芘上。可以從圖譜上還可看出苯環上-c-h(3100~3000cm-1)和c=c雙鍵(1591.27cm-1)的伸縮振動特征峰的出現。
通過圖2對1,3,6,8-4(4-甲酰基苯基)芘的結構進行紅外光譜分析。如圖2所示,結合1,3,6,8-四溴芘中的苯環(3100~3000cm-1)、苯環中c=c(1600~1580cm-1)、c-br(600-500cm-1)伸縮振動的特征峰位置,在1,3,6,8-4(4-甲酰基苯基)芘中可以看到苯環(3043.67cm-1)和苯環中c=c(1600.92cm-1)的伸縮振動特征峰仍然存在,且出現c=o醛基雙峰特征吸收峰(2723.49cm-1和2810.28cm-1)伸縮振動特征峰,同時,c-br伸縮振動的特征峰出現在513.07cm-1,但強度極弱,說明br幾乎完全被取代。
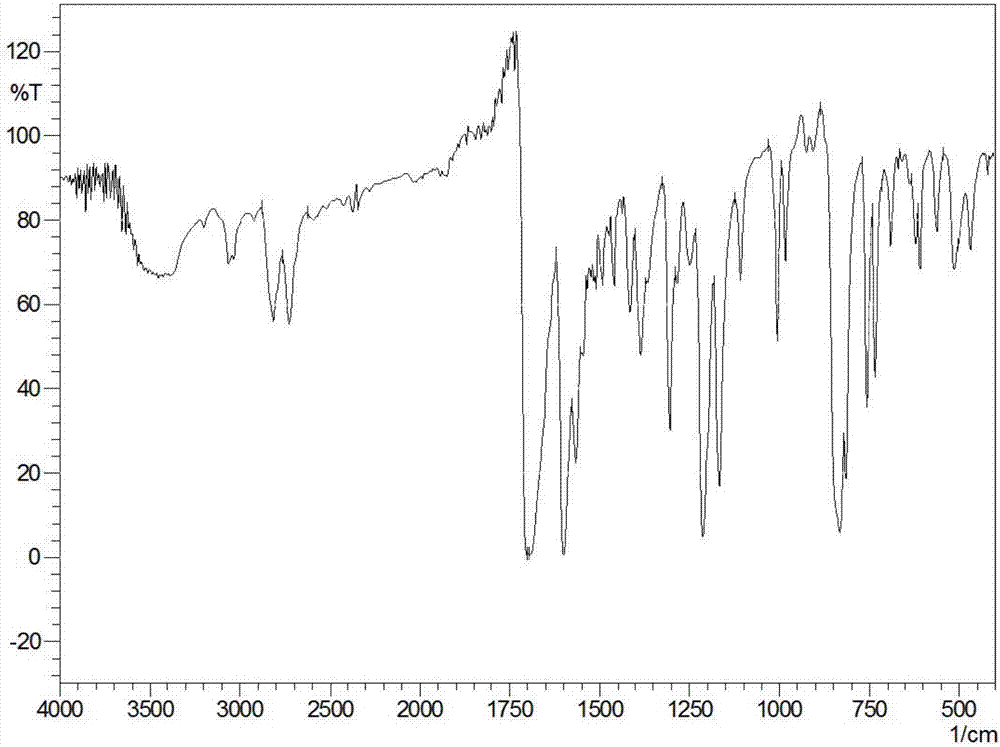
通過圖3的紅外圖譜,并結合1,3,6,8-4(4-甲酰基苯基)芘紅外圖譜對xwb-cof-1的結構進行分析,1,3,6,8-4(4-甲酰基苯基)芘上的c=o醛基雙峰特征吸收峰(2723.49cm-1和2810.28cm-1)在該圖譜中出現不明顯,而苯環上的特征峰-c-h(3100~3000cm-1)和c=c雙鍵(1604.77cm-1)的伸縮振動峰仍然存在,圖3中還出現了特征峰c-s(900~1300cm-1之間),峰值較弱。
圖4是有機骨架材料的熱重分析曲線。由圖4可知,a線是樣品質量隨著溫度變化的曲線,b線是熱流隨著溫度變化的曲線。開始加熱升溫后,樣品質量開始減小,這可能是因為樣品失水。樣品表面吸附的水的蒸發速率相對較快。到大約60℃時,樣品質量減小的速度開始變慢,說明此時水的蒸發變慢。樣品內部的水蒸發速率相對較慢。60至400℃之間樣品質量變化很慢,也說明了有機骨架相對較穩定。400℃以后有機骨架出現破壞。在約700℃之后產物的重量開始保持穩定,達到58%。證明得到的有機骨架材料結構穩定,熱穩定性能好。b線的變化也可以佐證上述變化過程。
圖5為本發明得到的有機骨架材料的10.0k的sem圖;圖6為本發明得到的有機骨架材料的5.0k的sem圖。通過電鏡圖片,可以看出該有機骨架材料在電鏡下呈現細長的棍棒狀,具有較高的比表面積。
- 還沒有人留言評論。精彩留言會獲得點贊!