一種微尺度下光誘導有機催化制備聚合物的方法與流程
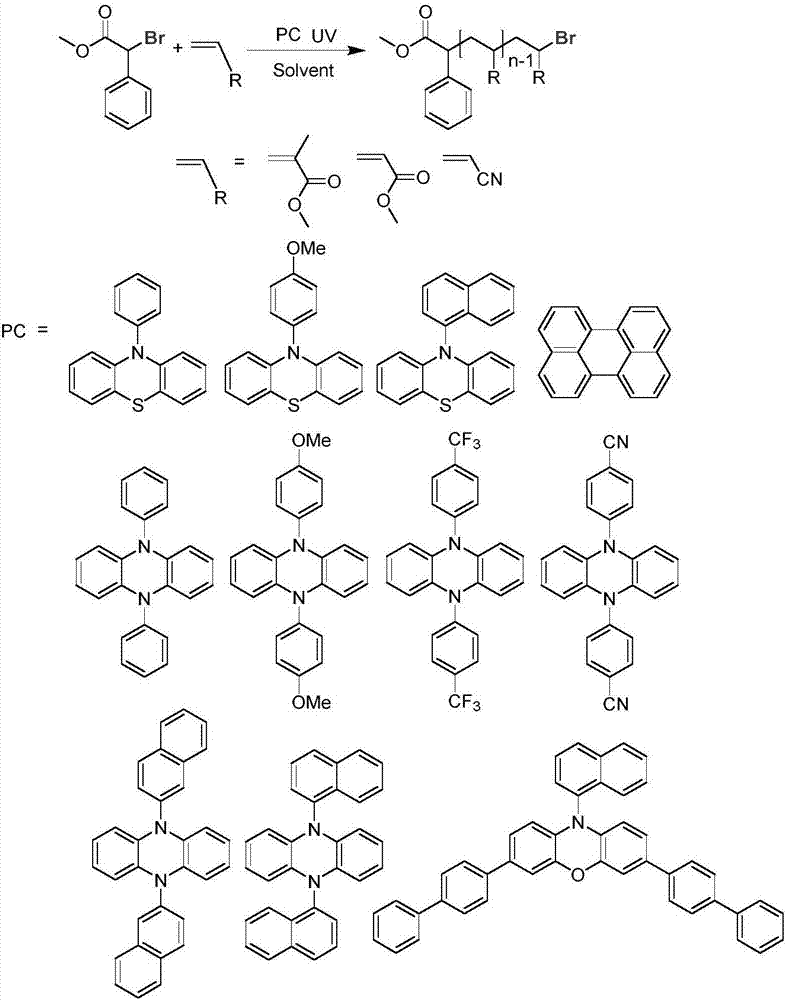
本發明屬于精細化學與高分子材料合成領域,具體涉及一種有機催化制備無金屬殘留聚合物聚(甲基)丙烯酸酯的方法。
背景技術:
聚(甲基)丙烯酸酯具有密度低、機械強度較強、熔點較低、透光率較高等優異的性能,這使它成為一種重要的聚合物。對其聚合方法的研究也比較多,其中活性自由基聚合(LRP)是生產聚(甲基)丙烯酸酯很好的聚合方法。活性自由基聚合(LRP)是進行分子設計、合成精確一級結構聚合物、實現對聚合物的分子量及分子量分布可控的重要途徑,已廣泛應用于功能高分子材料、聚合物刷的合成。而原子轉移自由基聚合(ATRP)是研究最為廣泛的活性自由基聚合(LRP)之一。(甲基)丙烯酸酯是常見的原子轉移自由基聚合單體。原子轉移自由基聚合(ATRP)通過一個以金屬催化劑[Cu(I),Ru(II),Fe(II)]進行調節的氧化還原過程進行聚合。使用金屬催化劑會使產物中有金屬殘留,從而限制了使用ATRP合成出的聚(甲基)丙烯酸酯在電學材料、生物材料等方面的應用。
技術實現要素:
本發明所要解決的技術問題是提供一種光誘導的有機催化制備聚合物的合成方法。以解決現有技術存在的引發劑效率低、反應速度慢、副反應多等缺陷。
為解決上述技術問題,本發明采取的技術方案是:
一種微尺度下光誘導有機催化制備聚合物的方法,,包括以下步驟:
(1)將有機催化劑溶解于溶劑中,得到第一均相溶液;
(2)將引發劑與單體混合均勻,得到第二均相溶液;
(3)將步驟(1)中所述第一均相溶液和步驟(2)中所述第二均相溶液分別同時泵入微反應裝置中的微混合器中,充分混合后通入微反應裝置中的微通道反應器中,光照下充分反應,收集產物。
步驟(1)中,所述的有機催化劑選自10-苯基吩噻嗪、10-(4-甲氧基苯基)吩噻嗪、10-(1-萘基)吩噻嗪、二萘嵌苯、3,7-二(4-(1,1'-聯苯))-(10-(1-萘基))-10-吩惡嗪、5,10-二苯基-5,10-二氫吩嗪、5,10-二(4-甲氧基苯基)-5,10-二氫吩嗪、5,10-二(4-(三氟甲基)苯基)-5,10-二氫吩嗪、5,10-二(4-(腈基)苯基)-5,10-二氫吩嗪、5,10-二(2-萘基)-5,10-二氫吩嗪或5,10-二(1-萘基)-5,10-二氫吩嗪中的一種或多種。
所述的有機催化劑的化學結構如下表:
步驟(1)中,所述溶劑選自二甲基亞砜、N,N-二甲基甲酰胺或N,N-二甲基苯胺中的一種或多種;所述的單體與溶劑體積比為0.2~2,優選為0.2~0.4。
步驟(2)中,所述的引發劑為烷基鹵素;優選的所述的引發劑為α-溴苯乙酸甲酯、2-溴丙酸甲酯或2-氯丙酸甲酯;
步驟(2)中,所述的單體選自甲基丙烯酸甲酯、丙烯酸甲酯或丙烯腈中的一種或多種;所述的引發劑和單體的摩爾比為1:50~200。
步驟(3)中,泵入所述微反應裝置中的第一均相溶液的流速為0.01mL/min~0.8mL/min,優選為0.03mL/min~0.06mL/min;第二均相溶液的流速為0.01mL/min~0.8mL/min,優選為0.012mL/min~0.095mL/min。
步驟(3)中,所述反應溫度為20℃~60℃,優選為20℃~40℃。
步驟(3)中,所述反應的滯留時間為15min~120min;
所述的光照的波長范圍為280nm~450nm。
步驟(3)中,所述微反應裝置包括第一進樣器、第二進樣器、微混合器、微通道反應器、接收器、光源;所述第一進樣器和第二進樣器并聯連接到微混合器;所述微混合器、微通道反應器和接收器串聯連接;所述光源位于微通道反應器外側,其光照范圍覆蓋微通道反應器;所述連接為通過管道連接。
優選的,所述微通道反應器為石英盤管,保留體積為1~20ml優選為5~20mL,管徑為0.2~2mm,優選為1~2mm;所述的第一進樣器和第二進樣器與微混合器之間的連接管長度為10cm~50cm,微混合器與微通道反應器之間的連接管長度為10cm~50cm,微通道反應器與接收器之間的連接管長度為10cm~50cm。
本發明的光照是在紫外光或可見光照射下進行的。
本發明將原子轉移自由基聚合與微流場技術相結合,建立微尺度下聚(甲基)丙烯酸酯高效合成的新方法,本發明大幅度提升物料混合、傳質和傳熱的效率,加快了反應速度;同時反應溫度分布更加均勻,光照更加均勻,連續流過程控制更加精確,能夠有效避免副產物的產生。
有益效果:本發明聚合反應速度快;轉化率高;副反應少,即單體自聚產生的均聚物含量低;同時得到的聚合物分子量分布窄。由于聚合過程中完全避免使用金屬催化劑,所得到的聚合物作為電學材料或生物材料具有極大的優勢。
附圖說明
圖1為本發明微通道反應器的結構示意圖。其中,1為第一進樣器,2為第二進樣器,3為微混合器,4為微通道反應器,5為接收器,6為光源。
圖2為α-溴苯乙酸甲酯引發乙烯類單體聚合的反應式。
具體實施方式
根據下述實施例,可以更好地理解本發明。然而,本領域的技術人員容易理解,實施例所描述的內容僅用于說明本發明,而不應當也不會限制權利要求書中所詳細描述的本發明。
本發明分別以α-溴苯乙酸甲酯、2-溴丙酸甲酯和2-氯丙酸甲酯為引發劑引發(甲基)丙烯酸酯進行有機催化原子轉移自由基聚合。通過變換單體,合成不同的無金屬殘留均聚物;通過調節引發劑與單體的配比,合成具有不同分子量的均聚物。
以下實施例中所述第一和第二進樣器使用雷弗注射泵TYD01,將反應液泵入微通道中進行聚合,實施例中使用的微反應器其保留體積為5ml,管徑為1mm。實施例中縮寫Mn表示聚合物數均分子量,PDI表示聚合物分子量分布指數,MMA表示甲基丙烯酸甲酯,MA表示丙烯酸甲酯,AN表示丙烯腈,DMSO表示二甲基亞砜。
所述有機催化劑的制備方法參考以下文獻,10-苯基吩噻嗪參考文獻[1]、10-(4-甲氧基苯基)吩噻嗪參考文獻[2]、10-(1-萘基)吩噻嗪參考文獻[3]、3,7-二(4-(1,1'-聯苯))-(10-(1-萘基))-10-吩惡嗪參考文獻[4]、5,10-二苯基-5,10-二氫吩嗪[5]、5,10-二(4-甲氧基苯基)-5,10-二氫吩嗪[6]、5,10-二(4-(三氟甲基)苯基)-5,10-二氫吩嗪、5,10-二(4-(腈基)苯基)-5,10-二氫吩嗪或5,10-二(1-萘基)-5,10-二氫吩嗪參考文獻[7]、5,10-二(2-萘基)-5,10-二氫吩嗪參考文獻[8]。本發明中制備得到的化合物結構和文獻報道的一致。此外二萘嵌苯可直接購買得到(CAS:198-55-0,純度:98%,生產廠家:北京百靈威科技有限公司)
[1]Eric B,Kochi K.Journal of the chemical society-perkin transactions,1995,8:1057-1064.
[2]Sunil K,Meenu S,Jwo-Huei J,et al.Journal of Materials Chemistry C:Materials for Optical and Electronic Devices,2016,4:6769-6777.
[3]Bu B P,Bong G S.Repub.Korean Kongkae Taeho Kongbo,2014,KR 2014076520A20140620.
[4]Pearson R M,Lim C,McCarthy B G,et al.J.Am.Chem.Soc.2016,138:11399-11407.
[5]Pokhodenko V D,Koshechko V G,Inozemtsev AN.Oxidation Communications,1986,8:141-7.
[6]Yoshigo S,Kiyoshi S,Shunkai S,et al.Jpn.Kokai Tokkyo Koho,1990,JP 02093466A 19900404.
[7]Theriot J C,Lim C,Yang H,et al.Science,2016,27:1082-1086.
[8]Chem-Hooi L,Matthew R,Blaine G,et al.Journal of the American Chemical Society,2017,139:348-355.
實施例1:
20℃下,在氮氣保護下將有機催化劑10-苯基吩噻嗪(31mg,0.1128mmol)溶解于溶劑DMSO(30ml)中,得到均相溶液后備用;將引發劑α-溴苯乙酸甲酯(345μl,2.256mmol)與單體MMA(12ml,112.8mmol)混合均勻后與上述均相溶液分別同時泵入微反應裝置中的微混合器中,控制DMSO流速為(0.059ml/min),MMA流速為(0.024ml/min),充分混合后通入有紫外光(380nm)照射的微通道反應器中,混合體系在微通道反應器中滯留時間為60min,流速為(0.083ml/min),充分反應,收集產物,用甲醇和水的混合液(1:1)沉淀。用乙醇清洗產物3次,以除去產物中殘留的單體和溶劑。將產物在30℃下真空干燥24h后,即為所要制備的聚甲基丙烯酸甲酯。轉化率為95%,Mn=5300g/mol,PDI=1.21。
實施例2:
20℃下,在氮氣保護下將有機催化劑10-苯基吩噻嗪(31mg,0.1128mmol)溶解于溶劑DMSO(30ml)中,得到均相溶液后備用;將引發劑2-溴丙酸甲酯(250μl,2.256mmol)與單體MMA(12ml,112.8mmol)混合均勻后與上述均相溶液分別同時泵入微反應裝置中的微混合器中,控制DMSO流速為(0.119ml/min),MMA流速為(0.048ml/min),充分混合后通入有紫外光(380nm)照射的微通道反應器中,混合體系在微通道反應器中滯留時間為30min,流速為(0.167ml/min),充分反應,收集產物,用甲醇和水的混合液(1:1)沉淀。用乙醇清洗產物3次,以除去產物中殘留的單體和溶劑。將產物在30℃下真空干燥24h后,即為所要制備的聚甲基丙烯酸甲酯。轉化率為94%,Mn=4900g/mol,PDI=1.37。
實施例3:
20℃下,在氮氣保護下將有機催化劑10-苯基吩噻嗪(31mg,0.1128mmol)溶解于溶劑DMSO(30ml)中,得到均相溶液后備用;將引發劑2-氯丙酸甲酯(245μl,2.256mmol)與單體MMA(12ml,112.8mmol)混合均勻后與上述均相溶液分別同時泵入微反應裝置中的微混合器中,控制DMSO流速為(0.238ml/min),MMA流速為(0.095ml/min),充分混合后通入有紫外光(380nm)照射的微通道反應器中,混合體系在微通道反應器中滯留時間為15min,流速為(0.333ml/min),充分反應,收集產物,用甲醇和水的混合液(1:1)沉淀。用乙醇清洗產物3次,以除去產物中殘留的單體和溶劑。將產物在30℃下真空干燥24h后,即為所要制備的聚甲基丙烯酸甲酯。轉化率為89%,Mn=5100g/mol,PDI=1.49。
實施例4:
30℃下,在氮氣保護下將有機催化劑10-(1-萘基)吩噻嗪(43.1mg,0.1324mmol)溶解于溶劑DMSO(30ml)中,得到均相溶液后備用;將引發劑α-溴苯乙酸甲酯(405μl,2.648mmol)與單體MA(12ml,132.4mmol)混合均勻后與上述均相溶液分別同時泵入微反應裝置中的微混合器中,控制DMSO流速為(0.059ml/min),MA流速為(0.024ml/min),充分混合后通入有紫外光(365nm)照射的微通道反應器中,混合體系在微通道反應器中滯留時間為60min,流速為(0.083ml/min),充分反應,收集產物,用甲醇和水的混合液(1:1)沉淀。用乙醇清洗產物3次,以除去產物中殘留的單體和溶劑。將產物在30℃下真空干燥24h后,即為所要制備的聚丙烯酸甲酯。轉化率為81%,Mn=3700g/mol,PDI=1.46。
實施例5:
30℃下,在氮氣保護下將有機催化劑二萘嵌苯(33.4mg,0.1324mmol)溶解于溶劑DMSO(30ml)中,得到均相溶液后備用;將引發劑α-溴苯乙酸甲酯(405μl,2.648mmol)與單體MA(12ml,132.4mmol)混合均勻后與上述均相溶液分別同時泵入微反應裝置中的微混合器中,控制DMSO流速為(0.030ml/min),MA流速為(0.012ml/min),充分混合后通入有可見光照射的微通道反應器中,混合體系在微通道反應器中滯留時間為120min,流速為(0.042ml/min),充分反應,收集產物,用甲醇和水的混合液(1:1)沉淀。用乙醇清洗產物3次,以除去產物中殘留的單體和溶劑。將產物在30℃下真空干燥24h后,即為所要制備的聚丙烯酸甲酯。轉化率為78%,Mn=3800g/mol,PDI=1.47。
實施例6:
40℃下,在氮氣保護下將有機催化劑3,7-二(4-(1,1'-聯苯))-(10-(1-萘基))-10-吩惡嗪(112.6mg,0.1834mmol)溶解于溶劑DMSO(30ml)中,得到均相溶液后備用;將引發劑α-溴苯乙酸甲酯(560μl,3.668mmol)與單體AN(12ml,183.4mmol)混合均勻后與上述均相溶液分別同時泵入微反應裝置中的微混合器中,控制DMSO流速為(0.039ml/min),AN流速為(0.016ml/min),充分混合后通入有可見光照射的微通道反應器中,混合體系在微通道反應器中滯留時間為90min,流速為(0.055ml/min),充分反應,收集產物,用甲醇和水的混合液(1:1)沉淀。用乙醇清洗產物3次,以除去產物中殘留的單體和溶劑。將產物在30℃下真空干燥24h后,即為所要制備的聚丙烯腈。轉化率為98%,Mn=3100g/mol,PDI=1.12。
實施例7:
40℃下,在氮氣保護下將有機催化劑5,10-二(1-萘基)-5,10-二氫吩嗪(79.7mg,0.1834mmol)溶解于溶劑DMSO(30ml)中,得到均相溶液后備用;將引發劑α-溴苯乙酸甲酯(560μl,3.668mmol)與單體AN(12ml,183.4mmol)混合均勻后與上述均相溶液分別同時泵入微反應裝置中的微混合器中,控制DMSO流速為(0.059ml/min),AN流速為(0.024ml/min),充分混合后通入有可見光照射的微通道反應器中,混合體系在微通道反應器中滯留時間為60min,流速為(0.083ml/min),充分反應,收集產物,用甲醇和水的混合液(1:1)沉淀。用乙醇清洗產物3次,以除去產物中殘留的單體和溶劑。將產物在30℃下真空干燥24h后,即為所要制備的聚丙烯腈。轉化率為96%,Mn=2900g/mol,PDI=1.22。
實施例8:
40℃下,在氮氣保護下將有機催化劑5,10-二(1-萘基)-5,10-二氫吩嗪(79.7mg,0.1834mmol)溶解于溶劑DMSO(60ml)中,得到均相溶液后備用;將引發劑α-溴苯乙酸甲酯(560μl,3.668mmol)與單體AN(12ml,183.4mmol)混合均勻后與上述均相溶液分別同時泵入微反應裝置中的微混合器中,控制DMSO流速為(0.118ml/min),AN流速為(0.024ml/min),充分混合后通入有可見光照射的微通道反應器中,混合體系在微通道反應器中滯留時間為60min,流速為(0.083ml/min),充分反應,收集產物,用甲醇和水的混合液(1:1)沉淀。用乙醇清洗產物3次,以除去產物中殘留的單體和溶劑。將產物在30℃下真空干燥24h后,即為所要制備的聚丙烯腈。轉化率為96%,Mn=2900g/mol,PDI=1.22。
比較例1:
向20ml無色透明玻璃瓶中加入聚四氟乙烯磁力攪拌子,加入10-苯基吩噻嗪(6.2mg,0.02256mmol),塞好橡膠塞,然后使用雙排管抽排三次除去瓶中的空氣。用錫箔紙包裹玻璃瓶使其完全避光。在氬氣保護的情況下加入DMSO(6ml),磁力攪拌5min。加入α-溴苯乙酸甲酯(69μl,0.4512mmol)和MMA(2.4ml,22.56mmol)。反應在磁力攪拌和365nm紫外燈照射下進行。反應結束后用甲醇和水的混合液(1:1)沉淀。用乙醇清洗產物3次,以除去產物中殘留單體和溶劑。將產物在30℃下真空干燥24h后,即為所要制備的聚甲基丙烯酸甲酯。轉化率為79%,Mn=4100g/mol,PDI=1.52。
- 還沒有人留言評論。精彩留言會獲得點贊!