一種2,6?二(4?吡啶乙烯)萘烷烴衍生物及其制備方法和應用與流程
本發明涉及一種萘烷烴衍生物及其制備方法和應用,特別是一種2,6-二(4-吡啶乙烯)萘烷烴衍生物及其制備方法和應用。
背景技術:
陰離子廣泛地存在于大自然中,在生態系統中承擔著最基礎的作用,陰離子的選擇性識別和檢測是當前超分子化學和分析化學領域內的一個重要研究課題。由于離子容易與水分子形成氫鍵而干擾識別測定,目前陰離子的檢測和識別一般都在無水條件下進行。而熒光探針的主要潛在應用領域是水體系中的生物、醫學和環境監測,所以在水溶液中的檢測才具有潛在的應用價值。本發明涉及一種對bf4-和pf6-具有識別功能的熒光試劑,能快速檢測識別bf4-和pf6-,且不受其他離子干擾。
熒光光譜法操作方便、不需預處理、對樣品沒有破壞、成本低和不受外界電磁場的影響等一系列的優點而受到越來越多的關注。設計合成對bf4-和pf6-具有選擇性的熒光探針,同時可實現對bf4-和pf6-的可視法檢測,這樣高效精確的檢測方法在醫學和生物學領域都有重要意義。
技術實現要素:
本發明的目的在于,提供一種2,6-二(4-吡啶乙烯)萘烷烴衍生物及其制備方法和應用。本發明能夠在中性水溶液中選擇性地檢測識別bf4-和pf6-,同時,操作簡單,測試結果可視,分析更直接。
本發明的技術方案:一種2,6-二(4-吡啶乙烯)萘烷烴衍生物,其結構式為如附圖1所示。
前述的2,6-二(4-吡啶乙烯)萘烷烴衍生物,所述結構式中,r的分子式為c7h15、c8h17或c9h19。
一種前述的2,6-二(4-吡啶乙烯)萘烷烴衍生物的制備方法,包括如下步驟:
(1)將2,6-二溴萘與4-吡啶乙烯、醋酸鈀、三苯基膦加入到事先通好氮氣的高壓反應釜中,加入三乙胺,在油浴中80~100℃下反應65~72小時;冷卻到室溫,加入二氯甲烷溶液,待固體全部溶解后,先用蒸餾水洗滌萃取,再用飽和食鹽水洗滌萃取;有機相洗滌完畢,加入無水硫酸鎂干燥20~30分鐘,過濾硫酸鎂固體;將溶液濃縮,出現大量灰黃色沉淀;過濾沉淀,用石油醚,正己烷洗滌沉淀,得到2,6-二(4-吡啶乙烯)萘純的產物;
(2)將2,6-二(4-吡啶乙烯)萘溶解于dmf中,分別加入1-溴代烷,在65~80℃,反應30~36小時,出現沉淀,冷卻至室溫,過濾沉淀,用少量的石油醚,環己烷和丙酮洗滌沉淀,即可得成品。
前述的2,6-二(4-吡啶乙烯)萘烷烴衍生物的制備方法,所述1-溴代烷為1-溴代庚烷、1-溴代辛烷或1-溴代壬烷。
一種前述的2,6-二(4-吡啶乙烯)萘烷烴衍生物在選擇識別bf4-和pf6-中的應用。
前述的2,6-二(4-吡啶乙烯)萘烷烴衍生物在選擇識別bf4-和pf6-中的應用,所述識別的方法是將2,6-二(4-吡啶乙烯)萘烷烴衍生物溶于甲醇,制得熒光試劑,然后向試劑中滴入待識別樣品,得樣品溶液,利用波長為415nm的激光對樣品溶液進行熒光激發,并檢測激發的熒光的最大發射波長即可。
前述的2,6-二(4-吡啶乙烯)萘烷烴衍生物在選擇識別bf4-和pf6-中的應用,所述2,6-二(4-吡啶乙烯)萘烷烴衍生物中,r的分子式為c7h15,當檢測到bf4-時,激發的熒光的最大發射波長為550nm;當檢測到pf6-時,激發的熒光的最大發射波長為590nm。
前述的2,6-二(4-吡啶乙烯)萘烷烴衍生物在選擇識別bf4-和pf6-中的應用,所述2,6-二(4-吡啶乙烯)萘烷烴衍生物中,r的分子式為c8h17,當檢測到bf4-時,激發的熒光的最大發射波長為600nm;當檢測到pf6-時,激發的熒光的最大發射波長為585nm。
前述的2,6-二(4-吡啶乙烯)萘烷烴衍生物在選擇識別bf4-和pf6-中的應用,所述2,6-二(4-吡啶乙烯)萘烷烴衍生物中,r的分子式為c9h19,當檢測到bf4-時,激發的熒光的最大發射波長為580nm;當檢測到pf6-時,激發的熒光的最大發射波長為590nm。
前述的2,6-二(4-吡啶乙烯)萘烷烴衍生物在選擇識別bf4-和pf6-中的應用,所述試劑中2,6-二(4-吡啶乙烯)萘烷烴衍生物的濃度為1.00×10-5mol·l-1。
本發明的有益效果:
1、本發明用于水溶液中bf4-和pf6-的定量分析,定量分析的線性濃度最低值為1.0×10-6mol·l-1。
2、其它共存常見的陰離子不干擾測定。
3、本發明操作簡單,測試結果可視,分析更直接。
為進一步說明本發明的有益效果,發明人做了如下實驗:
一、定性分析測試
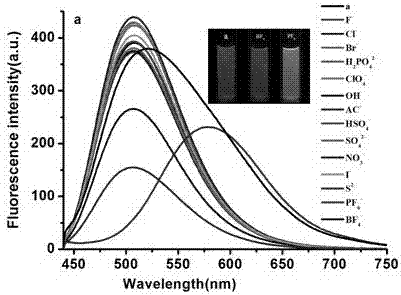
1、在濃度范圍為10-6~10-4mol/l的熒光探針水溶液(2,6-二(4-吡啶乙烯)萘烷烴衍生物水溶液)中,當激發波長為415nm時,熒光探針a(r為c7h15,下同)、b(r為c8h17,下同)、c(r為c9h19,下同)的最大發射波長是515nm,當熒光探針水溶液中存在bf4-混合物時,熒光探針的最大發射波長從515nm分別紅移至550nm、600n、580nm,熒光探針a由綠色變為淺綠色,探針b由藍綠色變為橘紅色,探針c藍綠色變為黃色,分別如圖2(a)、2(b)、2(c)所示。
2、在濃度范圍為10-6~10-4mol/l的熒光探針水溶液中,當激發波長為415nm時,熒光探針a、b、c的最大發射波長是515nm,當熒光探針水溶液中存在pf6-混合物時,熒光探針的最大發射波長從515nm分別紅移至590nm、585n、590nm,熒光探針a由藍色變為粉紅色,探針b由藍綠色變為黃色,探針c由藍綠色變為黃色如圖2所示。bf4-和pf6-的檢測限最低至10-6mol·l-1。
二、定量分析測試
1、分別稱取探針a(r為c7h15)、b(r為c8h17)、c(r為c9h19)質量為6.9mg、7.2mg、7.5mg,用甲醇溶于1.0ml容量瓶,濃度為10-2mol/l,根據需要用二次水逐級稀釋到10-4mol/l,得到探針a、b、c試劑。
2、稱取優級純的四丁基銨鹽配制成100ml的水溶液,陰離子濃度為10-2mol/l。根據需要用二次水逐級稀釋。
3、分別取5個10.0ml容量瓶中分別加入熒光探針1.00×10-4mol/l標準液1.0ml,分別加入1.00×10-4mol/l,0,0.2,0.5,1,2毫升的bf4-或pf6-溶液稀釋至刻度搖勻,室溫放置5分鐘;
4、引入熒光光譜進行測定,激發波長415nm。
5、以bf4-和pf6-濃度為橫坐標,熒光強度為縱坐標,得到工作曲線。
6、樣品測定,取10.0ml容量瓶,加入探針濃度為1.00×10-4mol/l的試劑1.0ml,加入被測bf4-和pf6-溶液,稀釋至刻度,室溫放置5分鐘,引入3.0cm的石英比色皿進行熒光測定,根據熒光強度在工作曲線上查出樣品濃度。檢測識別的最低濃度值為1.0×10-6mol·l-1。
三、其他陰離子熒光激發對比實驗
分別配制氨基酸濃度為1.00×10-2mol/l的且只含f-、cl-、br-、h2po42-、clo4-、oh-、ac-、hso4-、so42-、no3-、i-、s2-、pf6-、bf4-的水溶液,將水溶液分別利用本發明的方法(激發波長415nm)進行識別,結果如附圖2(a)、2(b)、2(c)所示,只有在識別只含bf4-和pf6-的時,熒光均發生紅移,而且在365nm紫外光燈照射下加入bf4-的探針a、b、c分別發出淺綠色,橘紅色,黃色。加入pf6-、的探針a、b、c分別發出粉紅色,黃色,黃色。
四、不同濃度的bf4-和pf6-的中性溶液激發測試
在探針a、b、c濃度為1.00×10-5mol/l,分別用bf4-和pf6-溶液滴定,隨著bf4-和pf6-的加入,a的515nm的熒光峰緩慢下降,達到某一程度時,bf4-離子在550nm左右出現扁平的峰,pf6-離子則在590nm處出現較矮的熒光峰;b的515nm的熒光峰緩慢下降,達到某一程度時,分別在600nm與585nm處緩慢出峰;隨著bf4-和pf6-的加入,c的515nm的熒光峰緩慢下降,達到某一程度時,分別在580nm與590nm處緩慢出峰(測試的激發波長為415nm),如圖3(a)、3(b)、3(c)所示。
五、抗干擾試驗
在探針濃度為1.00×10-5mol·l-1的試劑中,加入bf4-或pf6-后熒光發生紅移。再分別測試向a/b/c-bf4-或pf6-中加入其他f-、cl-、br-、h2po42-、clo4-、oh-、ac-、hso4-、so42-、no3-、i-、s2-、pf6-、bf4-后的熒光變化。
結果如圖4(a)、4(b)、4(c)所示,結果表明熒光試劑a、b、c檢測bf4-或pf6-的熒光強度,不受上述其他陰離子的影響。
附圖說明
圖1為探針a的結構式;
圖2(a)為熒光探針a與不同陰離子存在下的熒光光譜;
圖2(b)為熒光探針b與不同陰離子存在下的熒光光譜;
圖2(c)為熒光探針c與不同陰離子存在下的熒光光譜;
圖3(a)為不同濃度的pf6-對探針a的熒光滴定光譜圖;
圖3(b)為不同濃度的pf6-對探針b的熒光滴定光譜圖;
圖3(c)為不同濃度的pf6-對探針c的熒光滴定光譜圖;
圖4(a)為共存陰離子對探針a熒光法檢測pf6-的影響;
圖4(b)為共存陰離子對探針b熒光法檢測pf6-的影響;
圖4(c)為共存陰離子對探針c熒光法檢測pf6-的影響;
圖5(a)為r為c7h15時,熒光探針a的核磁譜圖;
圖5(b)為r為c8h17時,熒光探針b的核磁譜圖;
圖5(c)為r為c9h19時,熒光探針c的核磁譜圖。
具體實施方式
下面結合實施例對本發明作進一步的說明,但并不作為對本發明限制的依據。
本發明的實施例
實施例1、一種2,6-二(4-吡啶乙烯)萘烷烴衍生物,其結構式如附圖1所示;其中2,6-二(4-吡啶乙烯)萘烷烴衍生物的結構式中,r的分子式為c7h15,其核磁譜圖如附圖5(a)所示。
所述衍生物的制備方法包括如下步驟:
(1)將2mmol的2,6-二溴萘與5mmol的4-吡啶乙烯、醋酸鈀50mg、三苯基膦2mmol加入到事先通好氮氣的高壓反應釜中,加入5ml的三乙胺,在油浴中80℃下反應65小時;冷卻到室溫,加入250ml的二氯甲烷溶液,待固體全部溶解后,先用蒸餾水洗滌萃取,再用飽和食鹽水洗滌萃取;有機相洗滌完畢,加入無水硫酸鎂干燥20分鐘,過濾硫酸鎂固體;將溶液濃縮到1.5ml,出現大量灰黃色沉淀;過濾沉淀,用石油醚,正己烷洗滌沉淀,得到2,6-二(4-吡啶乙烯)萘純的產物;
(2)將1mmol的2,6-二(4-吡啶乙烯)萘溶解于2ml的dmf中,分別加入6mmol的1-溴代烷(1-溴代庚烷、1-溴代辛烷或1-溴代壬烷),在65℃,反應30小時,出現沉淀,冷卻至室溫,過濾沉淀,用少量的石油醚,環己烷和丙酮洗滌沉淀,即可得成品。
上述衍生物的應用方法是將其溶于甲醇后配制成濃度為1.00×10-5mol·l-1的溶液,然后想溶液中滴入待識別樣品,得樣品溶液,利用波長為415nm的激光對樣品溶液進行熒光激發;當檢測到bf4-時,激發的熒光的最大發射波長為550nm;當檢測到pf6-時,激發的熒光的最大發射波長為590nm。
實施例2、一種2,6-二(4-吡啶乙烯)萘烷烴衍生物,其結構式如附圖1所示;其中2,6-二(4-吡啶乙烯)萘烷烴衍生物的結構式中,r的分子式為c8h17,其核磁譜圖如附圖5(b)所示。
所述衍生物的制備方法包括如下步驟:
(1)將2mmol的2,6-二溴萘與5mmol的4-吡啶乙烯、醋酸鈀50mg、三苯基膦2mmol加入到事先通好氮氣的高壓反應釜中,加入5ml的三乙胺,在油浴中90℃下反應69小時;冷卻到室溫,加入250ml的二氯甲烷溶液,待固體全部溶解后,先用蒸餾水洗滌萃取,再用飽和食鹽水洗滌萃取;有機相洗滌完畢,加入無水硫酸鎂干燥25分鐘,過濾硫酸鎂固體;將溶液濃縮到1.5ml,出現大量灰黃色沉淀;過濾沉淀,用石油醚,正己烷洗滌沉淀,得到2,6-二(4-吡啶乙烯)萘純的產物;
(2)將1mmol的2,6-二(4-吡啶乙烯)萘溶解于2ml的dmf中,分別加入6mmol的1-溴代烷(1-溴代庚烷、1-溴代辛烷或1-溴代壬烷),在72℃,反應33小時,出現沉淀,冷卻至室溫,過濾沉淀,用少量的石油醚,環己烷和丙酮洗滌沉淀,即可得成品。
上述衍生物的應用方法是將其溶于甲醇后配制成濃度為1.00×10-5mol·l-1的溶液,然后想溶液中滴入待識別樣品,得樣品溶液,利用波長為415nm的激光對樣品溶液進行熒光激發;當檢測到bf4-時,激發的熒光的最大發射波長為600nm;當檢測到pf6-時,激發的熒光的最大發射波長為585nm。
實施例3、一種2,6-二(4-吡啶乙烯)萘烷烴衍生物,其結構式如附圖1所示;其中2,6-二(4-吡啶乙烯)萘烷烴衍生物的結構式中,r的分子式為c9h19,其核磁譜圖如附圖5(c)所示。
所述衍生物的制備方法包括如下步驟:
(1)將2mmol的2,6-二溴萘與5mmol的4-吡啶乙烯、醋酸鈀50mg、三苯基膦2mmol加入到事先通好氮氣的高壓反應釜中,加入5ml的三乙胺,在油浴中100℃下反應72小時;冷卻到室溫,加入250ml的二氯甲烷溶液,待固體全部溶解后,先用蒸餾水洗滌萃取,再用飽和食鹽水洗滌萃取;有機相洗滌完畢,加入無水硫酸鎂干燥30分鐘,過濾硫酸鎂固體;將溶液濃縮到1.5ml,出現大量灰黃色沉淀;過濾沉淀,用石油醚,正己烷洗滌沉淀,得到2,6-二(4-吡啶乙烯)萘純的產物;
(2)將1mmol的2,6-二(4-吡啶乙烯)萘溶解于2ml的dmf中,分別加入6mmol的1-溴代烷(1-溴代庚烷、1-溴代辛烷或1-溴代壬烷),在80℃,反應36小時,出現沉淀,冷卻至室溫,過濾沉淀,用少量的石油醚,環己烷和丙酮洗滌沉淀,即可得到純熒光探針a。
上述衍生物的應用方法是將其溶于甲醇后配制成濃度為1.00×10-5mol·l-1的溶液,然后想溶液中滴入待識別樣品,得樣品溶液,利用波長為415nm的激光對樣品溶液進行熒光激發;當檢測到bf4-時,激發的熒光的最大發射波長為580nm;當檢測到pf6-時,激發的熒光的最大發射波長為590nm。
- 還沒有人留言評論。精彩留言會獲得點贊!