一種構建耐鹽酵母菌株的新方法與流程
本發明屬于微生物學、分子生物學及食品發酵交叉領域,涉及基因工程技術領域,尤其涉及轉錄因子spt15重組質粒yeplac195-p-t-spt15的構建及其應用,尤其是一種構建耐鹽酵母菌株的新方法。
背景技術:
目前國內外針對spt15重組質粒yeplac195-p-t-spt15的構建及其應用,最主要的發現是其能通過大腸桿菌進行表達,而對于spt15重組質粒yeplac195-p-t-spt15能否在t酵母中進行表達,還尚未發現。
醬油是中國傳統的調味品,其發酵工藝包括低鹽固態和高鹽稀態兩種。而醬油的高鹽稀態發酵工藝的主要是需要加入耐鹽酵母(簡稱t酵母)來增強醬油的風味品質等。高鹽稀態發酵鹽水濃度要在16%以上,這就要求添加的酵母具有較高的耐鹽性,因此篩選耐高鹽的酵母菌株成為添加醬油風味的重要手段之一。
t酵母作為醬油發酵的主要菌株之一,有一個復雜的耐鹽系統,使得它在高鹽環境中保持正常的生理活動和新陳代謝的發酵。在過去二十年中,很多研究已經開始調查其對高鹽的適應機制,主要集中在它的等離子膜、atp酶和na+/h+轉運蛋白。鹽對酵母新陳代謝的影響開始被研究,例如在醬油生產過程中酯類揮發物的變化,細胞生長和在高鹽環境中糖的消耗。
通過檢索,發現如下一篇與本發明專利申請相關的專利公開文獻(以下簡稱對比文件1):
耐鹽耐酸酵母提取物及其制備方法和應用(cn104982875a),由活性酵母依次加入酵母抽提酶、中性蛋白酶、復合酶、核酸酶與風味酶,調節ph至4-10,同時升溫至30-90℃,酶解反應一段時間,然后升溫滅酶,再離心分離得到上清液即為酵母水解液,再進行美拉德反應制得的。包括如下步驟:(1)酵母水解液制備以活性酵母為原料,向其中加水進行攪拌均勻,然后依次加入酵母抽提酶、中性蛋白酶、復合酶、核酸酶與風味酶,調節ph至4-10,同時升溫至40-90℃,酶解反應一段時間,然后升溫滅酶,再離心分離得到上清液即為酵母水解液;(2)熱反應將步驟得到的酵母水解液與還原性單糖進行美拉德反應,即得耐鹽耐酸酵母提取物。
通過對比,本發明專利申請與上述專利公開文獻的耐鹽酵母菌株構建方法存在本質的不同。
技術實現要素:
本發明的目的是克服現有技術的不足之處,提供一種構建耐鹽酵母菌株的新方法,該方法擴大了對于轉錄因子spt15突變庫的研究范圍,成功地將含有spt15的yeplac195-p-t-spt15重組體質粒在t酵母尿嘧啶缺陷型菌株wm2中表達出來,得到一種構建耐鹽酵母菌株的新方法,將該方法所構建出的新的耐鹽t酵母菌株用于醬油發酵中,待發酵成熟后測定氨基酸態氮的含量,發現在發酵醬油的過程中能產生更多的氨基酸態氮,驗證其在醬油釀造中具有明顯優勢。
為實現上述目的,本發明采用的技術方案如下:
一種構建耐鹽酵母菌株的新方法,步驟如下:
⑴以野生型t酵母為出發菌株,得到具有遺傳穩定性的尿嘧啶缺陷型菌株wm2;
⑵構建spt15基因突變庫;
⑶構建spt15重組質粒yeplac195-p-t-spt15庫;
⑷將構建好的重組質粒yeplac195-p-t-spt15庫導入到w303中,選取優勢菌株提取質粒;
⑸將提取出的質粒導入到wm2中,即構建出新的耐鹽t酵母菌株。
而且,具體步驟如下:
⑴以野生型t酵母為出發菌株,得到具有遺傳穩定性的尿嘧啶缺陷型菌株wm2;
⑵同時,以釀酒酵母為模板,構建轉錄因子spt15基因突變庫;
⑶將啟動子基因連接到質粒yeplac195中,即得到質粒yeplac195-p;
⑷將終止子基因連接到步驟⑶中構建的質粒yeplac195-p中,即得到質粒yeplac195-p-t;
⑸將步驟⑵中構建的spt15基因突變庫連接到步驟⑷中質粒yeplac195-p-t中,即得到質粒yeplac195-p-t-spt15庫;
⑹將步驟⑸中構建的質粒yeplac195-p-t-spt15庫導入到w303中,選取優勢菌株提取質粒;
⑺從步驟⑹獲得的菌株中提取質粒,將該質粒導入到步驟⑴中構建的wm2中,即構建出新的耐鹽t酵母菌株。
而且,具體步驟如下:
⑴以野生型t酵母為出發菌株,利用最佳的紫外誘變條件對其進行誘變處理,最佳的紫外誘變條件為:在誘變劑甲基磺酸乙酯質量百分數為1%的條件下,取od660=1時期的t酵母,在紫外照射距離為25±1cm,對其進行誘變55min;
然后,在含鹽的尿嘧啶缺陷型平板和ypd平板上對經過紫外線照射處理后的t酵母進行涂布對照篩菌,將在尿嘧啶缺陷型平板上不能生長、在ypd平板上生長的菌株進行傳代處理,得到具有遺傳穩定性的尿嘧啶缺陷型耐鹽菌株wm2;
其中,所述含鹽的尿嘧啶缺陷型平板為:
不添加尿嘧啶的完全極限培養基,調節ph至7.5,115℃滅菌20min制得的平板;
⑵運用易錯pcr技術,在反應體系中,設置退火溫度為42-58℃,mg2+終濃度在1mm以上,對轉錄因子spt15進行大量擴增,收集得到含有spt15的大量易錯pcr產物,用乙醇醋酸鈉法純化后,得到spt15基因突變庫,-20℃存儲備用;
所用易錯pcr反應體系為:
⑶構建spt15重組質粒yeplac195-p-t-spt15庫;
具體步驟如下:
①使用sphi和hindⅲ雙酶消化步驟⑵中所收集的純化后的200bppcr產物以及質粒載體yeplac195,使用kpni和saci雙酶消化步驟⑵中所收集的純化后的400bppcr產物以及質粒載體yeplac195-p,使用psti和sali雙酶消化步驟⑵中所收集的純化后的720bppcr產物以及yeplac195-p-t,37℃酶切1-2h;
其中,所述200bppcr產物即為啟動子,其所使用的上引物為:5'-gcgagctctcggaatagctttaatggtg-3';下引物為:5'-gcggtaccaccgatgggaaatgactctt-3';所述400bppcr產物即為終止子,其所使用的上引物為:5'-gcgcatgcggaaggaattgaaccatata-3';下引物為:5'-gcaagcttgtttaaggaatccatcttta-3';所述720bppcr產物即為spt15基因片段;
②在80℃將限制性內切酶進行失活處理5min,將dna聚合酶連接反應體系放在22℃的培養箱或室溫下恒溫反應4h或過夜,通過連接得到重組質粒庫yeplac195-p-t-spt15;
⑷將構建好的重組質粒yeplac195-p-t-spt15庫導入到w303菌株中,并進行培養,在長出的菌落中隨機挑取數個菌株,從中篩選耐高鹽的優勢菌株,對耐鹽性好的菌株提取質粒;
⑸將提取出的質粒導入到步驟⑴中得到的尿嘧啶缺陷型菌株wm2中,通過培養做dilution驗證,從中篩選耐鹽性最高的菌株,即構建出新的耐鹽酵母菌株。
而且,:所述步驟⑸中使用電轉化方法將提取出的質粒導入到步驟⑴中得到的尿嘧啶缺陷型菌株wm2中。
而且,所述電轉化方法的電轉化條件為:dna質量為0.1μg,li+濃度60mm,電壓為1400v。
而且,所述步驟⑷的具體步驟如下:
將構建好的重組質粒yeplac195-p-t-spt15庫導入到w303菌株中,涂布在尿嘧啶營養缺陷型平板上,30℃倒置培養3-5天,待長出白色菌落后,隨機挑取長勢良好的菌落劃線轉接到含質量百分數2%鹽nacl的尿嘧啶營養缺陷型平板上,放于30℃培養箱中倒置培養3-5天,待長出菌落后,繼續挑取長勢旺盛的菌落劃線轉接到含質量百分數4%鹽nacl的尿嘧啶營養缺陷型平板上,經培養繼續劃線轉接到質量百分數6%鹽nacl的尿嘧啶營養缺陷型平板上,在含質量百分數6%鹽nacl的尿嘧啶營養缺陷型平板上隨機挑取長勢旺盛的單菌落,培養后提取質粒。
而且,所述步驟⑸中dilution驗證的具體步驟如下:
取培養過夜的菌液測od值,根據od值取菌數量相同的菌液量進行離心,離心條件為3000rpm,離心3min,之后用無菌水進行梯度稀釋,稀釋五個濃度梯度:1╳107、1╳106、1╳105、1╳104、1╳103,稀釋之后取1μl在平板中相同梯度的菌液一一對應進行點接,上述5個濃度均有菌生長即為耐鹽菌株。
本發明的優點和積極效果是:
1、本發明方法擴大了對于轉錄因子spt15突變庫的研究范圍,成功地將含有spt15的yeplac195-p-t-spt15重組體質粒在t酵母尿嘧啶缺陷型菌株wm2中表達出來,得到一種構建耐鹽酵母菌株的新方法,將該方法所構建出的新的耐鹽t酵母菌株用于醬油發酵中,待發酵成熟后測定氨基酸態氮的含量,發現在發酵醬油的過程中能產生更多的氨基酸態氮,驗證其在醬油釀造中具有明顯優勢。
2、本發明方法首次將質粒yeplac195-p-t-spt15在t酵母的尿嘧啶缺陷型菌株wm2中成功表達。
3、本發明方法將釀酒酵母的轉錄因子spt15通過突變后在wm2中表達,成功提高了wm2菌株的耐鹽性。
4、本發明方法最終構建的新耐鹽t酵母菌株在醬油發酵中,可以明顯提高醬油的重要理化指標氨基酸態氮的含量。
附圖說明
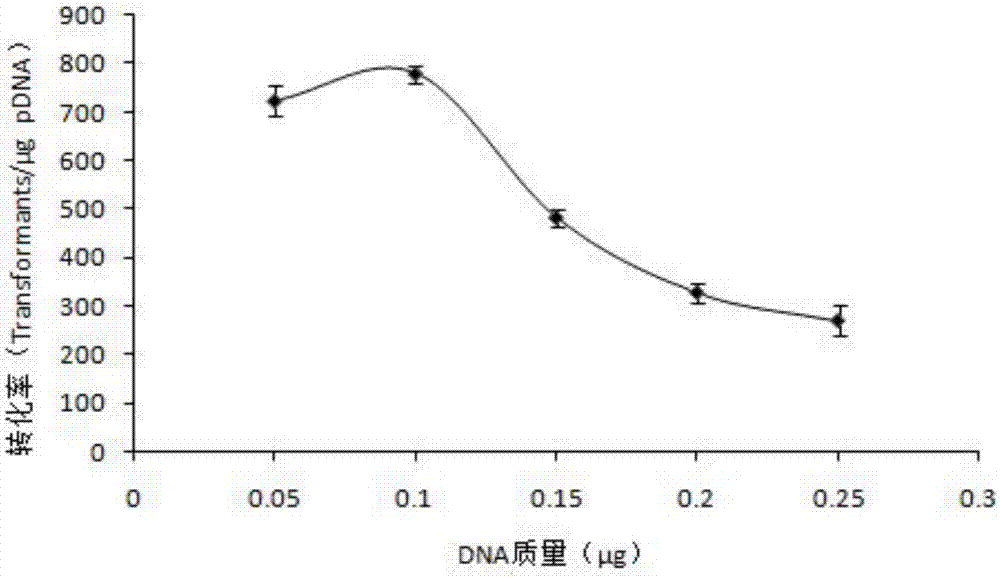
圖1為本發明中dna質量對t1菌株電轉化效率的影響圖;
圖2為本發明中li+濃度對t1菌株電轉化效率的影響圖;
圖3為本發明中電壓對t1菌株電轉化效率的影響圖;
圖4為本發明中重組質粒yeplac195-p-t-spt15構建過程圖;
圖5為本發明中氨基酸態氮的測量結果圖;
圖6為本發明中提取yeplac195-p-t-spt15質粒pcr產物電泳圖。
具體實施方式
下面結合實施例,對本發明進一步說明;下述實施例是說明性的,不是限定性的,不能以下述實施例來限定本發明的保護范圍。
本發明中所使用的原料,如無特殊說明,均為常規的市售產品;本發明中所使用的方法,如無特殊說明,均為本領域的常規方法。
一種構建耐鹽酵母菌株的新方法,步驟如下:
⑴以野生型t酵母為出發菌株,得到具有遺傳穩定性的尿嘧啶缺陷型菌株wm2;
⑵構建spt15基因突變庫;
⑶構建spt15重組質粒yeplac195-p-t-spt15庫;
⑷將構建好的重組質粒yeplac195-p-t-spt15庫導入到w303中,選取優勢菌株提取質粒;
⑸將提取出的質粒導入到wm2中,即構建出新的耐鹽t酵母菌株。
具體地,步驟如下:
⑴以野生型t酵母為出發菌株,得到具有遺傳穩定性的尿嘧啶缺陷型菌株wm2;
⑵同時,以釀酒酵母為模板,構建轉錄因子spt15基因突變庫;
⑶將啟動子基因連接到質粒yeplac195中,即得到質粒yeplac195-p;
⑷將終止子基因連接到(3)中構建的質粒yeplac195-p中,即得到質粒yeplac195-p-t;
⑸將(2)中構建的spt15基因突變庫連接到(4)中,即得到質粒yeplac195-p-t-spt15庫;
(6)將(5)中構建的質粒yeplac195-p-t-spt15庫導入到w303中,選取優勢菌株提取質粒;
(7)從(6)獲得的菌株中提取質粒,將該質粒導入到(1)中構建的wm2中,即構建出新的耐鹽t酵母菌株。
更具體地,步驟如下:
⑴構建t酵母尿嘧啶營養缺陷型菌種wm2
以野生型t酵母為出發菌株,利用最佳的紫外誘變條件對其進行誘變處理,最佳的紫外誘變條件為:在誘變劑甲基磺酸乙酯(ems)質量濃度為1%的條件下,取od660=1時期的t酵母,在紫外照射距離為25±1cm,對其進行誘變55min;
然后,在尿嘧啶缺陷型培養基平板(即為不添加尿嘧啶的完全極限培養基,調節ph至7.5,115℃滅菌20min制得的平板)和ypd平板上對經過紫外線照射處理后的t酵母進行涂布對照篩菌,將在尿嘧啶缺陷型平板上不能生長、在ypd平板上生長的菌株進行傳代處理,得到具有遺傳穩定性的尿嘧啶缺陷型耐鹽菌株wm2;
⑵利用易錯pcr構建spt15基因突變庫
運用易錯pcr技術對轉錄因子spt15進行大量擴增,得到spt15基因突變庫;
易錯pcr技術是在正常pcr技術的基礎上演變而來的,基于易錯pcr技術可運用于基因的隨機誘變。本發明正是通過改變pcr反應體系的條件和低保真的taqdna聚合酶,降低pcr過程中dna復制的保真度,以增加dna子鏈的堿基錯配率,得到較多點突變的擴增產物。本發明中運用易錯pcr技術對轉錄因子spt15進行大量擴增,得到spt15基因突變庫。
本實驗采用單因素試驗,分別在溫度、mg2+濃度、datp、dttp、dctp、dgtp的強度及濃度方面對pcr反應進行優化,找出最優點(即退火溫度為42-58℃,mg2+終濃度在1mm以上,datp、dttp、dctp、dgtp添加量的質量比例為4:1:1:4),從而發現:
①pcr反應在退火溫度為42-58℃溫度范圍內pcr產物濃度較高,在其他溫度范圍內則容易得到錯配率較高的易錯pcr產物,為隨機突變提供了有利的條件。
②提高體系中mg2+的量時可以得到錯配率高的易錯pcr產物。
③改變pcr體系中datp和dgtp的添加量較容易得到易錯pcr產物,對基因的誘變率較高;改變dttp和dctp的量不容易得到易錯pcr產物,所得基因的誘變率不高。
隨后在pcr熱循環儀中按表1所示反應體系進行pcr擴增,設置退火溫度為42-58℃,mg2+終濃度在1mm以上,收集得到含有spt15的大量易錯pcr產物,用乙醇醋酸鈉法純化后放-20℃存儲備用;
表1本發明所用易錯pcr反應體系表
⑶構建spt15重組質粒yeplac195-p-t-spt15庫;
經雙酶切、dna聚合酶連接等操作構建spt15重組質粒庫,具體步驟如下:
①使用sphi和hindⅲ雙酶消化步驟⑵中所收集的純化后的200bp(啟動子)pcr產物(上引物:5'-gcgagctctcggaatagctttaatggtg-3';下引物:5'-gcggtaccaccgatgggaaatgactctt-3')以及質粒載體yeplac195,使用kpni和saci雙酶消化步驟⑵中所收集的純化后的400bp(終止子)pcr產物(上引物:5'-gcgcatgcggaaggaattgaaccatata-3';下引物:5'-gcaagcttgtttaaggaatccatcttta-3')以及質粒載體yeplac195-p,使用psti和sali雙酶消化步驟⑵中所收集的純化后的720bp(spt15基因片段)pcr產物以及yeplac195-p-t,37℃酶切1-2h;
②在80℃將限制性內切酶進行失活處理5min,將dna聚合酶連接反應體系,放在22℃的培養箱或室溫下恒溫反應4h或過夜,通過連接得到重組質粒庫yeplac195-p-t-spt15。構建步驟如圖4所示。
⑷將構建好的重組質粒yeplac195-p-t-spt15庫導入到w303菌株中,并進行培養,在長出的菌落中隨機挑取數個菌株,從中篩選耐高鹽的優勢菌株,對耐鹽性好的菌株提取質粒;
更具體地,步驟如下:
將構建好的重組質粒yeplac195-p-t-spt15庫導入到w303菌株中,涂布在尿嘧啶營養缺陷型平板上,30℃倒置培養3-5天,待長出白色菌落后,隨機挑取長勢良好的菌落劃線轉接到含質量百分數2%鹽nacl的尿嘧啶營養缺陷型平板上,放于30℃培養箱中倒置培養3-5天,待長出菌落后,繼續挑取長勢旺盛的菌落劃線轉接到含質量百分數4%鹽nacl的尿嘧啶營養缺陷型平板上,經培養繼續劃線轉接到質量百分數6%鹽nacl的尿嘧啶營養缺陷型平板上;
在含6%鹽nacl的尿嘧啶營養缺陷型平板上隨機挑取長勢旺盛的4個單菌落,分別命名為303-1,303-2,303-3,303-4。培養后提取質粒,分別命名為a1,a2,a3,a4。
⑸將提取出的質粒導入到步驟⑴中得到的尿嘧啶缺陷型菌株wm2中,即構建出新的耐鹽酵母菌株;
將空白質粒yep195、a1、a2、a3、a4質粒分別導入wm2菌株,得到菌株a0、a1、a2、a3、a4。將這5株菌進行醬油發酵,測定其重要理化指標氨基酸態氮含量,結果如圖5所示。由圖5中可知a1,a2,a3,a4菌株比a0菌株在發酵醬油的過程中能產生更多的氨基酸態氮。
同時,通過培養在不同鹽濃度的尿嘧啶缺陷型平板上做dilution驗證(取培養過夜的菌液測od值,根據od值取菌數量相同的菌液量進行離心3000rpm,3min,之后用無菌水進行梯度稀釋,稀釋五個濃度梯度(1╳107、1╳106、1╳105、1╳104、1╳103),稀釋之后取1μl在平板中相同梯度的菌液一一對應進行點接,上述5個濃度均有菌生長即為耐鹽菌株,經dilution實驗可以看出四個質粒在耐鹽方面有一定的優勢。
將得到的a1,a2,a3,a4質粒進行pcr反應,看是否有目的條帶,并將所得的pcr產物進行測序。所得pcr產物的電泳條帶見圖6。經基因測序分析,得到的a1,a2,a3,a4此四個基因是所需要的目的基因。
較佳地,所述步驟⑸中使用電轉化方法將提取出的質粒導入到步驟⑴中得到的尿嘧啶缺陷型菌株wm2中。
電轉化法是酵母轉化的比較通用的方法,真菌轉化方法中,常用的ca2+轉化法、冷凍貯藏法、peg法、傳統醋酸鋰轉化法對于真菌種類的覆蓋率均不如電轉化方法。因此,本發明選擇電轉化法對t酵母進行轉化,同時對電轉化方法進行優化,確定了最佳電轉化方法。
①dna質量對t1菌株電轉化效率的影響
pzeu質粒濃度及純度:od260/od280=1.70,od260=0.28,dna濃度=14μg/ml。
1μgdna需要加入71.4μldna溶液(0.1μg=7.1μl,0.3μg=21.4μl,0.5μg=35.7μl,0.7μg=50.0μl,0.9μg=64.3μl)。
采用已有標準方法的條件,dna質量=0.1μg時轉化子78個,轉化率=780transformants/μgpdna。0.1μgdna即飽和,對于t酵母來說,高濃度的dna反而使轉化效率顯著降低,最高轉化效率的產生仍然來源于少量dna,如圖1所示。
②li+濃度對t1菌株電轉化效率的影響
li+濃度對t1轉化效率的影響較為明顯,最優轉化濃度仍然是60mm,對應轉化率為492個/μgpdna,含有147個轉化子,如圖2所示。
③電壓對t1菌株電轉化效率的影響
電壓對t1酵母電轉化率的影響為線性增長,1500v以前轉化率線性提升,最高效電壓<1500v,轉化率為491transformants/μgpdna(此電壓在0.1cm電轉槽較容易產生電火花,0.2cm電轉槽轉入成功率比較高。),如圖3所示。
以上可得:t酵母最適電轉化條件為:dna質量為0.1μg,li+濃度60mm,電壓約1400v的時候。
以上所述,僅是本發明的較佳實施例而已,并非對本發明做其它形式的限制,任何技術人員可能利用上述揭示的技術內容加以變更或改型為等同變化的等效實施例;但是凡是未脫離本發明的技術方案內容,對上述實施例做任何簡單修改、等同變化與改型,仍屬于本發明技術方案的保護范圍。
sequencelisting
<110>天津科技大學
<120>一種構建耐鹽酵母菌株的新方法
<130>2017-3-7
<160>6
<170>patentinversion3.3
<210>1
<211>28
<212>dna
<213>spt15上引物
<400>1
gcgtcgacatggctgacgagggacgttt28
<210>2
<211>28
<212>dna
<213>spt15下引物
<400>2
gcctgcagctacattttcctgaattcac28
<210>3
<211>28
<212>dna
<213>200bppcr產物上引物
<400>3
gcgagctctcggaatagctttaatggtg28
<210>4
<211>28
<212>dna
<213>200bppcr產物下引物
<400>4
gcggtaccaccgatgggaaatgactctt28
<210>5
<211>28
<212>dna
<213>400bppcr產物上引物
<400>5
gcgcatgcggaaggaattgaaccatata28
<210>6
<211>28
<212>dna
<213>400bppcr產物下引物
<400>6
gcaagcttgtttaaggaatccatcttta28
- 還沒有人留言評論。精彩留言會獲得點贊!