一種二氧化錳催化芳烴或雜環芳烴三氟甲基化的方法與流程
本發明屬于催化有機合成技術領域,具體涉及一種MnO2催化芳烴或雜環芳烴三氟甲基化的方法。
背景技術:
由于均相催化劑在使用時存在催化劑難以回收、藥劑費用高、引入雜質等問題,所以非均相催化劑的使用備受關注。
二氧化錳是一種顏色為棕黑色或黑色的金屬錳化合物,是處中間價態的正四價,同時具有氧化性和還原性兩重性質,而且在堿性條件下容易被氧化成六價錳,在酸性條件下則可以還原成二價錳。二氧化錳是一種環境友好廉價易得的過渡金屬氧化物,具有獨特的物理和化學性質,使它在材料領域占有重要的地位,在催化反應,電化學,分子吸附,磁學,光學,生物傳感器等領域展示了廣闊的應用前景。
三氟甲基(-CF3)具有獨特的化學性能及生物活性,將其引入到有機化合物中可以顯著地改變該化合物的偶極矩、極性、親脂性以及化學和代謝穩定性。因此不斷尋求有效的三氟甲基化合物合成方法,成為氟化學領域研究的一個熱點。其中,通過產生三氟甲基自由基形成碳碳鍵是三氟甲基化的重要策略。考慮二氧化錳的催化特性,試圖將二氧化錳引入到三氟甲基化反應中。目前將二氧化錳應用到三氟甲基化反應的不多,該文章中二氧化錳體系也不是主要反應體系。我們以二氧化錳為催化劑,實現了對芳烴及雜環化合物的三氟甲基化,并對其機理進行了深入的探究。
由于氟原子較小的原子尺寸與強的電負性,向有機分子中引入氟原子可以顯著改變分子的物理化學性質和生物化學性質,如增大分子的極性,提高分子的脂溶性,增強分子在生物體新陳代謝過程中的穩定性等。據統計,目前有30%的農業化學品和20%的藥物中至少含有一個氟原子。天然的含氟有機物極少,大多數的含氟有機物需要人為引入氟原子。三氟甲基化作為一種向有機分子中引入氟原子的有效方式被廣泛應用于醫藥,農業化學品等的生產。含三氟甲基的藥物有很多,如用來治療關節炎的新藥塞來昔布,抗艾滋藥物依法韋侖,治療糖尿病的特效藥捷諾維等。
工業上生產三氟甲苯常用的方法是Swarts 方法,即先將甲苯氯化得到三氯甲苯,再用路易斯酸三氟化銻或者氫氟酸將三氯甲苯氟化得到三氟甲芳烴或雜環芳烴。該方法步驟繁多,環境污染大。近些年來,苯的三氟甲基化研究有了較大發展。按照反應的底物不同,可以將反應方法大致分為兩類,第一種是C-X鍵的三氟甲基化(X=Cl、Br、I、B(OH)2),第二種是C-H鍵的直接三氟甲基化。雖然以Cu、Ag、Pd等催化劑為代表的C-X三氟甲基化反應體系具有選擇性好,產率高的優點,但是需要預先對C-H鍵進行功能化,使其轉化成C-X鍵。從簡化反應步驟,綠色化學,原子經濟性等角度,直接C-H鍵的三氟甲基化更符合現代化學的要求。
技術實現要素:
本發明的目的在于提供一種MnO2催化芳烴或雜環芳烴三氟甲基化的有機合成方法,其體系簡單、反應條件溫和、成本低,解決了目前工業上制備存在的高污染、高成本的問題。
為實現上述目的,本發明采用如下技術方案:
一種MnO2催化芳烴或雜環芳烴三氟甲基化的方法,是以二氧化錳作為催化劑,三氟甲烷亞磺酸鈉(CF3SO2Na-Langlois試劑)作為三氟甲基的來源,乙腈為溶劑,在低溫常壓條件下催化芳烴或雜環芳烴三氟甲基化。
其具體包括如下步驟:
1)將0.1~0.2mmol二氧化錳和0.2~0.5mmol三氟甲烷亞磺酸鈉于反應器中混合后,加入1~5mL乙腈和0.5~1mL芳烴或雜環芳烴;
2)將該反應器放入超聲波清洗機中超聲處理30s后,將反應器連接空氣氣球,置于集熱式恒溫磁力攪拌器中50℃反應24h。
所述超聲處理的頻率為40-60KHz。
本發明的優點在于:
(1)本發明采用二氧化錳為催化劑進行低溫催化,使芳烴或雜環芳烴的三氟甲基化反應在較溫和、較低的溫度(<60℃)條件下即可進行,且反應后的二氧化錳環境友好廉價易得,不僅降低了成本,也更加環保。
(2)本發明采用非均相反應體系,反應條件溫和,可有效避免目前工業三氟甲基化反應中使用強酸、重金屬帶來的污染。
(3)本發明方法操作簡單易行,具有很好的重復性,且反應過程簡單、對環境友好,有利于大規模的工業生產,適于推廣應用。
(4)本發明方法可用于對具有生物活性、藥物活性的有機化合物進行三氟甲基修飾,具有良好的實際應用前景和經濟效益。
附圖說明
圖1為實施例1反應后分離測得三氟甲苯純品19F NMR圖譜。
圖2為實施例2三氟甲烷亞磺酸鈉熱重測試。
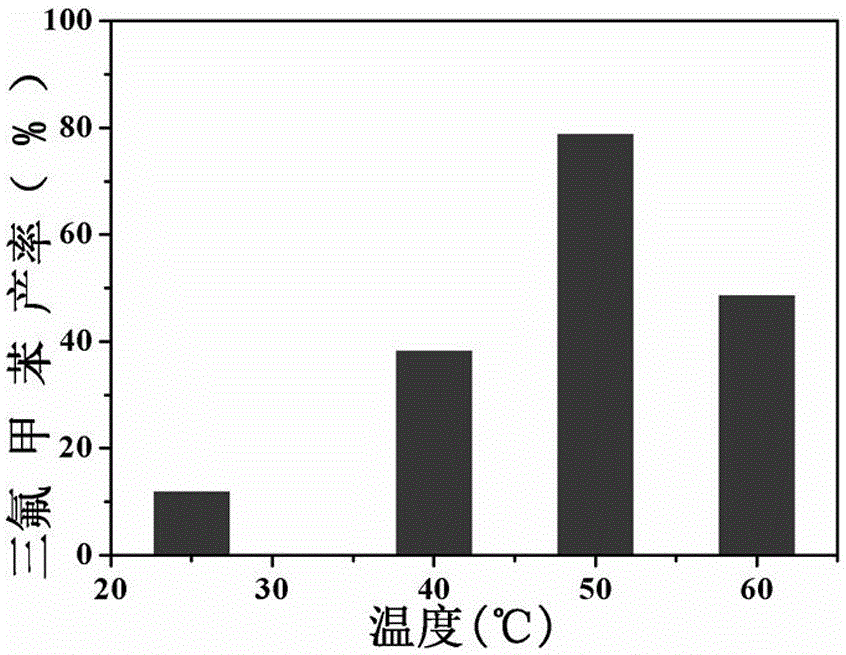
圖3為實施例3以底物苯為例不同溫度對三氟甲苯產率的變化關系圖。
圖4為實施例4以底物苯為例不同反應時間與三氟甲苯產率的變化關系圖。
圖5為實施例5所得以三氟甲氧基苯為內標物測得三氟甲芳烴或雜環芳烴的核磁19F NMR產率。
圖6為實施例6反應過程中捕獲的中間產物三氟甲基自由基的ESR圖。
具體實施方式
為了使本發明所述的內容更加便于理解,下面結合具體實施方式對本發明所述的技術方案做進一步的說明,但是本發明不僅限于此。
實施例1(底物以苯為例)
(1)稱取0.2mmol二氧化錳作為催化劑,0.5mmol三氟甲烷亞磺酸鈉作為三氟甲基源,于反應器中混合后,加入1mL乙腈為溶劑及0.5mL反應物苯,封上封口膜;
(2)將該反應器放入超聲波清洗機中50KHz超聲處理30s后,將反應器連接空氣氣球,置于集熱式恒溫磁力攪拌器中50℃反應24h;
(3)反應完全后離心,將上清液過柱分離得到純凈的三氟甲苯后測得19F NMR圖譜(見圖1)。
實施例2(底物以苯為例)
(1)稱取0.2mmol二氧化錳作為催化劑,0.5mmol三氟甲烷亞磺酸鈉作為三氟甲基源,于反應器中混合后,加入1mL乙腈為溶劑及0.5mL苯,封上封口膜;
(2)將該反應器放入超聲波清洗機中50KHz超聲處理30s后,將反應器連接空氣氣球,置于集熱式恒溫磁力攪拌器中反應24h,溫度分別為25℃,40℃,50℃,60℃;
(3)反應完全后離心,將上清液用GC(產物量已標定)檢測;
結果顯示(見圖2),隨著反應溫度升高產率逐漸升高,但并不是無限的升高,在50℃達到最高。
實施例3
(1)分別稱取10-20mg三氟甲烷亞磺酸鈉記為1,2,在空氣和氮氣氛圍下進行熱重測試結果見圖3中的曲線1,2;
(2)稱取10mg三氟甲烷亞磺酸鈉和10mg二氧化錳的混合物在空氣氛圍下進行熱重測試,結果見圖3中的曲線3;
(3)測試結果表明三氟甲基源三氟甲烷亞磺酸鈉的分解溫度為150-190℃遠大于反應所需要的溫度50℃。
實施例4(底物以苯為例)
(1)稱取0.2mmol二氧化錳作為催化劑,0.5mmol三氟甲烷亞磺酸鈉作為三氟甲基源,于反應器中混合后,加入1mL乙腈為溶劑及0.5mL底物苯,封上封口膜;
(2)將該反應器放入超聲波清洗機中50KHz超聲處理30s后,將反應器連接空氣氣球,置于集熱式恒溫磁力攪拌器中50℃分別反應6、12、18、24h。
由不同反應時間與三氟甲苯產率的變化關系圖(見圖4)可以看出,隨反應時間的延長,三氟甲苯產率提高。
實施例5
(1)稱取0.2mmol二氧化錳作為催化劑,0.5mmol三氟甲烷亞磺酸鈉作為三氟甲基源,于反應器中混合后,加入1mL乙腈為溶劑及0.5mL反應物芳烴和雜環芳烴,封上封口膜;
(2)將該反應器放入超聲波清洗機中50KHz超聲處理30s后,將反應器連接空氣氣球,置于集熱式恒溫磁力攪拌器中50℃反應24h;
(3)反應完全后離心,取0.7mL上清液和10uL三氟甲氧基苯(內標)混合均勻后加入到核磁管中,測得各產物的19F NMR圖譜如圖5,得到氟譜產率。
實施例6
(1)稱取0.1mmol二氧化錳作為催化劑,0.2mmol三氟甲烷亞磺酸鈉作為三氟甲基源,于離心管中混合后,加入1mL乙腈,并標為A;
(2)稱取1mg 2-甲基-2-亞硝基丙烷二聚物(MNP)于離心管中,加入1mL乙腈,并標為B;
(3)取0.5mL B于A中,加熱2h后由電子順磁共振(ESR)測定三氟甲基的自由基,結果見圖6。
以上所述僅為本發明的較佳實施例,凡依本發明申請專利范圍所做的均等變化與修飾,皆應屬本發明的涵蓋范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 含離子液體共溶劑介質中制備(R)?3,5?雙三氟甲基苯乙醇的方法與流程
- 一種鄰二氟苯的合成方法與制造工藝
- 2,2,2?三氟乙硫醇的合成的制造方法與工藝
- 2-氯-3-(甲基硫烷基)-N-(1-甲基-1H-四唑-5-基)-4-(三氟甲基)苯甲酰胺或其鹽在耐受HPPD抑制劑除草劑的轉基因作物植物的區域中防治不想要的植物的用途的制造方法與工藝
- (R)?3,5?雙(三氟甲基)苯乙醇的生物制備方法與制造工藝
- 2-(2-吡啶)-6-(2-氯-3-三氟甲基苯甲酰基)-5,7,8-三氫吡啶并[4,3-d]嘧啶及其制備方法與制造工藝
- 一種1,3,4-噻二唑類化合物及其制備方法與制造工藝
- 一種1,3,4-噻二唑類化合物及其制備方法與制造工藝
- 一種阿瑞吡坦中間體及其制備方法與制造工藝
- 一類含4,4’?二氟二苯甲基的不對稱α?二亞胺鎳配合物、其中間體、制備方法及其應用與制造工藝