一種基于咪唑磺酸的金屬有機框架及制備方法與應用與流程
本發明屬于納米材料制備技術領域,具體涉及一種基于咪唑磺酸的金屬有機框架及制備方法與應用。
背景技術:
金屬有機框架(metal-organic frameworks,MOFs)材料是將有機配體和金屬離子通過自組裝形成的具有重復網絡結構的一種雜化材料,是近幾十年來配位化學領域中發展得比較快的新材料。與傳統的無機多孔材料相比,MOFs材料具有更大的空隙率和比表面積,尤其是可調節的孔徑以及可變的功能基團,因而MOFs材料作為多孔的功能化材料在氣體分離和存儲方面的應用表現出很大的優勢。目前,MOFs材料已經應用于氫氣存儲、藥物運載、催化反應、生物傳感器、氣體吸附與分離等方面。金屬有機框架材料的研究涉及有機化學、無機化學、配位化學、材料化學、生命科學以及計算機科學等學科的最新成果,因而近年來MOFs受到越來越多研究團隊的關注。
現有的金屬有機框架材料作為催化劑使用,其催化中心多限于其本身的不飽和金屬位點,具體地,由于二甲基甲酰胺(DMF)、水、乙醇等小溶劑分子的存在,未飽和的金屬中心與其進行結合來滿足配位需求,經過加熱或真空處理后可以去除這些溶劑分子,從而使不飽和金屬位點暴露。在移除軸向配體后露出的金屬位點可作為路易斯酸催化中心來加速反應的進行。然而,這種采用現有的金屬有機框架材料作為催化劑,由于僅依靠金屬位點作為路易斯酸的催化中心,使得催化效果欠佳。同時,有機強酸類分子催化劑能夠高效催化縮合、酯化等反應。然而,采用這種分子催化劑不利于催化劑的回收以及產物的分離。
技術實現要素:
為克服現有技術的缺陷,本發明提供了一種基于咪唑磺酸的金屬有機框架,其將強酸型離子液體固定到金屬有機框架材料上,大大提高了金屬有機框架的催化效果,并且采用該金屬有機框架進行催化能夠多次回收利用。
為了將強酸型離子液體能夠固定在金屬有機框架上,本發明提供了用于合成基于咪唑磺酸的金屬有機框架的有機配體L,其化學結構式為:
命名為2-(1-咪唑基)-對苯二甲酸
為了能夠獲得上述有機配體L,本發明還提供了有機配體L的合成方法。首先,將2-甲基-對苯二甲酸進行反應獲得中間體A(即2-甲基-對苯二甲酸二酯),其次,以中間體A和溴代琥珀酰亞胺為原料進行反應得到中間體B,再次,以中間體B和咪唑為原料進行反應得到中間體C,最后中間體C經過水解反應即得有機配體L。
其中,中間體A的結構式為:
其中,R為碳原子數小于3的烷基,例如甲基、乙基、丙基等。
中間體B的結構式為:
其中,R為碳原子數小于3的烷基,例如甲基、乙基、丙基等。
中間體C的結構式為:
其中,R為碳原子數小于3的烷基,例如甲基、乙基、丙基等。
為了制備基于咪唑磺酸的金屬有機框架,本發明的還提供了基于咪唑磺酸的金屬有機框架的前體,即基于咪唑配體的金屬有機框架,其化學結構式為[Zr6O4(OH)4L6]n,其中,L為上述有機配體L,n為大于0的自然數。
本發明提供的一種基于咪唑磺酸的金屬有機框架,采用磺酸基化合物對上述基于咪唑配體的金屬有機框架進行合成修飾從而獲得。
其合成簡式如下:
其中,為基于咪唑配體的金屬有機框架的結構簡式,
為基于咪唑磺酸的金屬有機框架的結構簡式,R1為烷基或H原子被F原子取代的烷基。
本發明還提供了一種上述基于咪唑磺酸的金屬有機框架在納米催化中的應用。采用基于咪唑磺酸的金屬有機框架作為催化劑,反應條件溫和,反應時間短,催化劑用量少,可回收重復利用。
為了降低苯甲醛的縮合反應條件,本發明提供了一種苯甲醛縮合反應方法,以苯甲醛和乙二醇為底物,以上述基于咪唑磺酸的金屬有機框架為催化劑,在90℃、1atm下,以甲苯為溶劑進行縮合反應。
本發明的有益效果:
(1)本發明將強酸型聚離子液體的特征結構引入到金屬有機框架納米多孔材料中,拓寬了聚離子液體材料的種類,并實現了兩種材料的功能集成。
(2)本發明中的基于咪唑磺酸的金屬有機框架在常壓下對苯甲醛催化效果明顯,具有反應條件相對溫和,反應時間較短,催化劑用量少,可回收和重復利用的特點。
附圖說明
構成本申請的一部分的說明書附圖用來提供對本申請的進一步理解,本申請的示意性實施例及其說明用于解釋本申請,并不構成對本申請的不當限定。
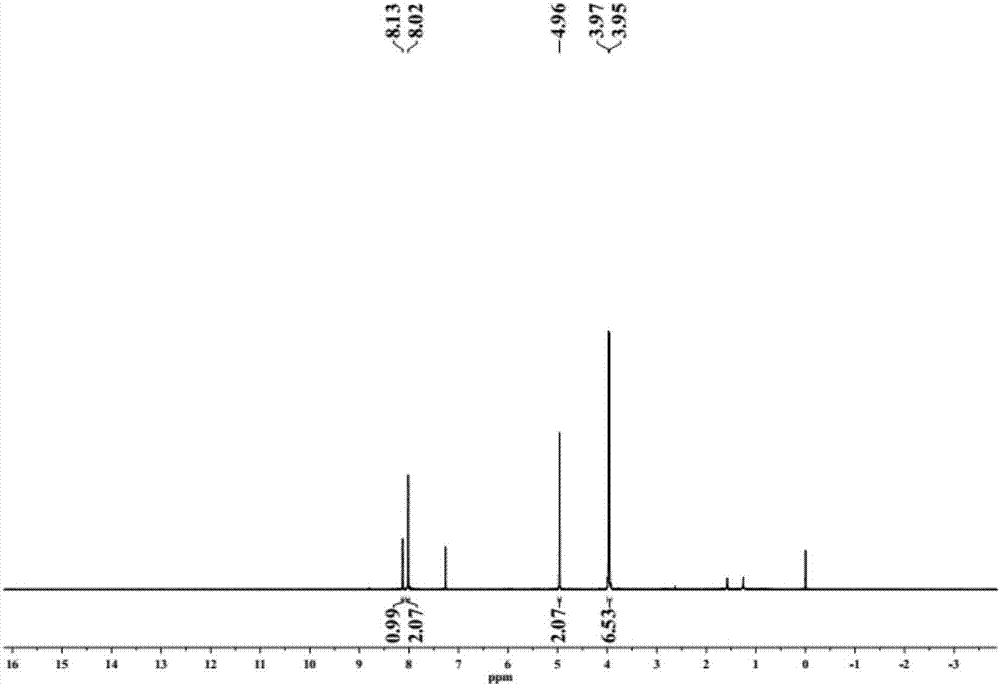
圖1為中間體A的1H-NMR譜圖;
圖2為中間體B的1H-NMR譜圖;
圖3為中間體C的1H-NMR譜圖;
圖4為有機配體L的1H-NMR譜圖;
圖5為中間體C直接與丙基磺酸內酯反應的產物D的1H-NMR譜圖;
圖6為基于咪唑配體的金屬有機框架的SEM圖;
圖7為基于咪唑配體的金屬有機框架的PXRD譜圖;
圖8為消解后的基于咪唑配體的金屬有機框架的1H-NMR譜圖;
圖9為基于咪唑配體的金屬有機框架在77K下N2吸附曲線;
圖10為基于咪唑磺酸的金屬有機框架的PXRD譜圖;
圖11為基于咪唑配體的金屬有機框架后合成修飾的紅外譜圖;
圖12為消解后的基于咪唑磺酸的金屬有機框架的1H-NMR譜圖;
圖13為監測基于咪唑磺酸的金屬有機框架催化縮合反應產率曲線;
圖14為基于咪唑磺酸的金屬有機框架重復5次催化縮合反應產率譜圖;
圖15為重復催化5次的基于咪唑磺酸的金屬有機框架的PXRD譜圖。
具體實施方式
應該指出,以下詳細說明都是例示性的,旨在對本申請提供進一步的說明。除非另有指明,本文使用的所有技術和科學術語具有與本申請所屬技術領域的普通技術人員通常理解的相同含義。
需要注意的是,這里所使用的術語僅是為了描述具體實施方式,而非意圖限制根據本申請的示例性實施方式。如在這里所使用的,除非上下文另外明確指出,否則單數形式也意圖包括復數形式,此外,還應當理解的是,當在本說明書中使用術語“包含”和/或“包括”時,其指明存在特征、步驟、操作、器件、組件和/或它們的組合。
本發明中所述合成修飾是指將磺酸基化合物與金屬有機框架進行合成,使得金屬有機框架被磺酸基化合物修飾的過程。
本發明中所述的1H-NMR譜圖為核磁共振氫譜譜圖,其中,1H-NMR為核磁共振氫譜的英文簡稱。
本發明中所述的SEM圖為掃描電子顯微鏡照片,其中,SEM為掃描電子顯微鏡(簡稱掃描電鏡)的英文簡稱。
本發明中所述的PXRD譜圖為多晶粉末X射線衍射譜圖,其中,PXRD多晶粉末X射線衍射的英文簡稱。
正如背景技術所介紹的,現有技術中存在的金屬有機框架材料無法固定強酸型離子液體的不足,為了解決如上的技術問題,本申請提出了一種用于合成基于咪唑磺酸的金屬有機框架的有機配體L。
本申請的一種典型實施方式中,提供了一種用于合成基于咪唑磺酸的金屬有機框架的有機配體L,其名稱為2-(1-咪唑基)-對苯二甲酸,其化學結構式為:
該有機配體中不僅含有能夠與金屬離子形成配位鍵的羧基,通過兩個羧基能夠使該有機配體與金屬離心形成金屬有機框架,而且該有機配體中的咪唑基團中叔胺氮能夠固定強酸型離子液體,從而使含有該有機配體L的金屬有機框架能夠固定強酸型離子液體。
為了能夠獲得上述有機配體L,本發明還提供了有機配體L的合成方法。首先,將2-甲基-對苯二甲酸進行反應獲得中間體A(即2-甲基-對苯二甲酸二酯),其次,以中間體A和溴代琥珀酰亞胺為原料進行反應得到中間體B,再次,以中間體B和咪唑為原料進行反應得到中間體C,最后中間體C經過水解反應即得有機配體L。
其中,中間體A的結構式為:
其中,R為碳原子數小于3的烷基,例如甲基、乙基、丙基等。
中間體B的結構式為:
其中,R為碳原子數小于3的烷基,例如甲基、乙基、丙基等。
中間體C的結構式為:
其中,R為碳原子數小于3的烷基,例如甲基、乙基、丙基等。
上述中間體A的反應可以通過2-甲基-對苯二甲酸與醇經過酯化反應一步合成中間體A,也可以先將2-甲基-對苯二甲酸與鹵代試劑(如三氯化磷、五氯化磷、亞硫酰氯等)反應,使3-甲基-對苯二甲酸中的羧基轉化成酰鹵,然后再加入醇進行醇解反應得到中間體A。由于酯化反應步驟較少,時間短,易于操作,適于實驗室中的研究應用。而先經過酰鹵再經過醇解的雖然步驟較多,但是其轉化率、產品收率較高,能夠降低生產成本,因而更適于工業化生產。
上述所述醇是指含有脂肪烴、脂環烴或芳香烴側鏈中的氫原子被羥基取代而成的化合物。由于上述合成中間體A的反應涉及酯化反應和醇解反應,碳鏈較少的低級醇為液態,有利于反應的進行,因而優選的醇為甲醇、乙醇或丙醇。
上述酯化反應的步驟為,向3-甲基-對苯二甲酸中加入甲醇和濃硫酸,以濃硫酸為催化劑,加熱回流反應后,調節pH至中性,抽濾、洗滌后即得中間體A。
優選的,所述中間體B的合成步驟為,以所述中間體A和溴代琥珀酰亞胺為原料,加入引發劑,加熱回流后經純化得到中間體B。
所述純化為將產品進行提純的過程。
進一步優選的,所述純化的步驟為,將回流后的液體進行冷卻,減壓蒸餾后獲得粗產品,然后對粗產品進行柱層析分離得到中間體B。
所述粗產品為純度較低的產品。經柱層析分離的產品純度大于99%。
所述引發劑為有機過氧化物引發劑或偶氮類引發劑(例如偶氮二異丁腈)等。
所述中間體A、溴代琥珀酰亞胺和引發劑的摩爾比為1:(1~1.5):(0.1~0.2)。
優選的,以中間體B和咪唑為原料進行的反應需在乙腈中進行。乙腈對中間體B及咪唑的溶解性更好,能夠使中間體B和咪唑充分接觸,從而提高反應速率及反應物的轉化率。
優選的,所述中間體C的合成反應的條件為加熱至80℃,回流2小時。
對中間體C的純化過程為:將回流反應后的液體進行減壓蒸餾除,再進行重結晶后即得中間體C。
優選的,所述中間體B和咪唑的摩爾比為1:(1.2~2)。由于有機合成反應均為可逆反應,其物料的轉化率基本不可能達到100%,提高咪唑的投入摩爾量能夠極大的提高中間體B的轉化率。從而極大的減少由于分步合成產生原料及能源的損失。
本發明所述水解反應為酯的水解。
優選的,所述水解反應的步驟為,分別加入中間體C、氫氧化鋰及溶劑,在室溫下攪拌后,調節pH,減壓蒸餾去除溶劑后即得有機配體L。
進一步優選的,所述溶劑為甲醇與水的混合物。其中,甲醇與水的體積比為3:1。
進一步優選的,攪拌的時間為10~14h。保證水解反應的充分進行。
進一步優選的,所述調節pH為采用氫溴酸調節pH=2~3。
進一步優選的,中間體C與氫氧化鋰的摩爾比為1:10。
為了制備基于咪唑磺酸的金屬有機框架,本發明的還提供了基于咪唑磺酸的金屬有機框架的前體,即基于咪唑配體的金屬有機框架,其化學結構式為[Zr6O4(OH)4L6]n,其中,L為上述有機配體L,n為大于0的自然數。
為了合成基于咪唑配體的金屬有機框架,本發明還提供了基于咪唑配體的金屬有機框架的制備方法,將上述有機配體L、酸和鋯鹽溶于極性溶劑,加熱至120±5℃,保持溫度24~48h,反應后即得金屬有機框架。
本發明中,所述酸可以為有機酸,如冰醋酸、苯甲酸等,優選冰醋酸;也可以為無機酸,如鹽酸等。
優選的,所述極性溶劑為極性強、介電常數大的溶劑。例如N,N-二甲基甲酰胺(DMF)、甲醇、乙醇、乙醚等。優先DMF。
優選的,所述鋯鹽為四氯化鋯、四溴化鋯、四碘化鋯及硝酸鋯。優選四氯化鋯。
優選的,反應后需將溫度降至室溫。
本發明中所述的室溫為15~25℃。
優選的,有機配體L、鋯鹽、酸和極性溶劑加入量的比為0.01:0.01:0.3:(3~5),mol:mol:mol:L。
具體的,以四氯化鋯為例,其合成簡式如下:
本發明提供的一種基于咪唑磺酸的金屬有機框架,采用磺酸基化合物對上述基于咪唑配體的金屬有機框架進行合成修飾從而獲得。
其合成簡式如下:
其中,為基于咪唑配體的金屬有機框架的結構簡式,
為基于咪唑磺酸的金屬有機框架的結構簡式,R1為烷基或H原子被F原子取代的烷基。
優選的,所述合成修飾的步驟為將基于咪唑配體的金屬有機框架置于非極性溶劑中加入帶有磺酸基的化合物50℃±5℃攪拌24h~36h,在60℃±5℃下真空干燥12h~24h。
本發明中,所述磺酸基的化合物可以為三氟甲磺酸,甲基磺酸,磺酸內酯等,優選丙基磺酸內酯。
優選的,所述非極性溶劑為極性弱、介電常數小的溶劑。例如丙酮,二氯甲烷,氯仿,苯,四氯化碳等。優先丙酮。
本發明還提供了一種上述基于咪唑磺酸的金屬有機框架在納米催化中的應用。采用基于咪唑磺酸的金屬有機框架作為催化劑,反應條件溫和,反應時間短,催化劑用量少,可回收重復利用。
為了提高上述基于咪唑磺酸的金屬有機框架的催化效果,本發明提供一種上述基于咪唑磺酸的金屬有機框架的活化方法,其具體步驟為,將金屬有機框架置于無水乙醇中浸泡活化24~48h,在60℃下真空干燥12h~24h。
優選的,所述活化的方法為,將金屬有機框架置于無水乙醇中浸泡活化36h,在60℃下真空干燥12h。
為了降低苯甲醛的縮合反應條件,本發明提供了一種苯甲醛縮合反應方法,以苯甲醛和乙二醇為底物,以上述基于咪唑磺酸的金屬有機框架為催化劑,在90℃、1atm下,以甲苯為溶劑進行縮合反應。
“atm”為atmosphere的簡寫,指地球上海平面的標準大氣壓,本發明中“atm”為壓強單位,1[atm]=1.01325Bar=760mmHg。
為了使得本領域技術人員能夠更加清楚地了解本申請的技術方案,以下將結合具體的實施例與對比例詳細說明本申請的技術方案。
實施例1:有機配體L的制備
(1)將2-甲基-對苯二甲酸(0.9g,5mmol)與甲醇(50mL)混合,在濃硫酸(5mL)的催化下反應,回流12小時。冷卻,抽濾得到粗產品為白色固體,即為中間體A,產率為95.0%,中間體A的1H-NMR譜圖見圖1。
(2)在100mL圓底燒瓶中先加入2-甲基-對苯二甲酸二甲酯(1.04g,5mmol),溴代琥珀酰亞胺(1.335g,7.5mmol),AIBN(0.246g,1.5mmol),再加入苯(45mL),加熱至80℃,回流12小時。冷卻,減壓蒸除溶劑,得粗產品。用二氯甲烷溶解,過濾得濾液,減壓除去溶劑,柱層析分離(石油醚:二氯甲烷=1:1)得淺黃色半固態1.45g,即為中間體B,產率為45.0%,中間體B的1H-NMR譜圖見圖2。
(3)在氮氣保護下,100ml的三口圓底燒瓶中加入咪唑(0.136g,2mmol),氫化鈉(0.048g,2mmol),四氫呋喃(45mL),加熱至65℃,回流2小時,再加入中間體B(0.285g,1mmol),回流2.5小時,冷卻,減壓蒸除溶劑,得產品,黃色油狀物,即為中間體C,產率為85%,中間體C的1H-NMR譜圖見圖3。
(4)在100mL的圓底燒瓶中加入中間體C,氫氧化鋰(10equiv),甲醇:水(=3:1),室溫下攪拌12h,冷卻,氫溴酸調節pH=2~3,減壓蒸除溶劑,白色產物析出,即為有機配體L,產率為90%,有機配體L的1H-NMR譜圖見圖4。
實施例2:配體與磺酸化合物的反應性
將實施例1中得到的中間體C(0.274g,1mmol)與等摩爾量的丙基磺酸內酯(0.112g,1mmol)在30mL丙酮中回流5h,旋蒸除去溶劑,獲得產物D的1H-NMR譜圖見圖5。
實施例3:基于咪唑配體的金屬有機框架的制備
將ZrCl4(28.8mg,0.12mmol)和冰醋酸(0.342mL),通過超聲處理(20分鐘)溶解在4.8mL的DMF中。然后將有機配體L(29.52mg,0.12mmol)加入到高壓釜中,并將該溶液超聲處理10~30min。然后將混合物加熱至120℃條件下維持24~48h。冷卻至室溫,然后進行離心,得到白色結晶粉末,依次用新鮮DMF(~10-20毫升×3)、有機溶劑(~10-20毫升×3)進行洗滌,離心,然后在40~80℃下真空干燥,即得粉末狀金屬有機框架晶體材料,SEM照片見圖6,PXRD譜圖見圖7,消解后晶體的1H-NMR譜圖見圖8。
其中,“~10-20毫升×3”表示為大約10-20毫升洗滌3次。
實施例4:金屬有機框架的氣體吸附性能
將實施例3中的金屬有機框架置于無水乙醇中浸泡活化48h,在60℃下真空干燥12h,對其進行氣體吸附測試:200mg的樣品置于預稱重的樣品管中,在120℃下脫氣10h,然后進行氣體的吸附與脫附測試,測試77K下的N2吸附曲線,吸附數據見圖9。
實施例5:基于咪唑磺酸的金屬有機框架的制備
將實施例3中得到的金屬有機框架晶體材料與1,3-丙基磺酸內酯(20μL),于丙酮(10mL)中回流,離心,然后在40~80℃下真空干燥,即得后修飾后的粉末狀金屬有機框架晶體材料,PXRD譜圖見圖10,反應前后的紅外譜圖見圖11(1190cm-1處出現了磺酸基的特征吸收峰),消解后晶體的1H-NMR譜圖見圖12。
實施例6:基于咪唑磺酸的金屬有機框架的金屬有機框架的催化性能
將實施例5中的基于咪唑磺酸的金屬有機框架置于無水乙醇中浸泡活化48h,在60℃下真空干燥12h,在10mL的圓底燒瓶中,加入甲苯(2mL),苯甲醛(20μL),乙二醇(120μL),后修飾的咪唑鹽配體L的金屬有機框架(1.0wt.%),90℃,回流,分別按時間15min、30min、45min、60min、90min、120min、180min、240min取樣,進行氣相表征,其表征結果見圖13,產率可達99.9%。將實施例5中的金屬有機框架再置于無水乙醇中浸泡活化48h,在60℃下真空干燥12h,重復反應五次,催化效果依然明顯,其表征結果見圖14,產率均在96%以上。通過對重復催化5次的基于咪唑磺酸的金屬有機框架進行PXRD表征,其結果顯示框架結構依然完好,結果見圖15。
實施例7:基于咪唑配體的金屬有機框架的催化性能
將實施例3中的基于咪唑配體的金屬有機框架置于無水乙醇中浸泡活化48h,在60℃下真空干燥12h,在10mL的圓底燒瓶中,加入甲苯(2mL),苯甲醛(20μL),乙二醇(120μL),基于咪唑配體的金屬有機框架(1.0wt.%),90℃,回流,反應4h后進行氣相表征,產率僅為58%。
上述雖然結合附圖對本發明的具體實施方式進行了描述,但并非對發明保護范圍的限制,所屬領域技術人員應該明白,在本發明的技術方案的基礎上,本領域技術人員不需要付出創造性勞動即可做出的各種修改或變形仍在本發明的保護范圍內。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 1?(5,6?二氯?1h?苯并[d]咪唑?2?基)?1h?吡唑?4?羧酸的葡甲胺鹽制劑的制作方法
- 一種多苯并咪唑類配合物及其制備方法和用圖
- 一種對氨基苯磺酰咪唑及其制備方法
- 苯并咪唑酸性鹽類化合物及其在催化合成混合二元酸二甲酯中的應用
- Salviskinone A的苯并咪唑基和二(2-甲硫基乙基)胺基衍生物的組合物在抗缺氧藥物中 ...的制作方法
- Salviskinone A的苯并咪唑基和二(2-甲硫基乙基)胺基衍生物的組合物在預防或治療胰 ...的制作方法
- Salviskinone A的苯并咪唑基和二(2-甲硫基乙基)胺基衍生物的組合物在抗菌藥物中的應用
- 一種含咔唑、苯并咪唑取代的喹啉衍生物及其制備方法和應用
- 一種3,7-二叔丁基-s-(三氟甲基)二苯并噻吩三氟甲磺酸鹽的工業生產方法
- 一種晶型的苯并咪唑基喹啉亞銅配合物發光材料的制作方法