利用報告基因選擇性富集突變核酸片段及其方法與流程
本發明屬于分子生物學領域,涉及基因工程和基因分析,具體涉及一種利用報告基因選擇性富集突變核酸片段的方法。
背景技術:
腫瘤是一種病因尚未完全闡明的疾病,主要有基因突變所導致。因此,篩選腫瘤高危人群以及進行早期診斷和治療尤為重要。近年來,隨著免疫學,生物化學,分子生物學及相應技術的發展,腫瘤生物學標志物的研究也得到了深化和突破,尤其是腫瘤循環dna的發現,為腫瘤早期診斷的無創性和敏感性帶來了契機。并使循環dna成為腫瘤早期診斷研究的新靶點。
自殺基因(suicidegene),是指將某些病毒或細菌的基因導入靶細胞中,其表達的酶可催化無毒的藥物前體轉變為細胞毒物質,從而導致攜帶該基因的受體細胞被殺死,此類基因稱為自殺基因。自殺基因若能正常表達,其攜帶的基因表達的蛋白質便能殺死宿主細胞,若自殺基因那段序列不能正常編碼即移碼或含有終止密碼子,便不能正常表達自殺基因,宿主細胞便能正常生長。利用熱點突變如kras12位點突變,突變之后含有終止密碼子,將這段序列克隆到自殺基因中,看他在自殺基因上的生長情況。若突變前,不含有終止密碼子,便不能正常在宿主細胞上生長;突變后,含有終止密碼子,能在宿主細胞上生長。自殺基因具有敏感性高,假陽性等優點,從而用于檢測循環dna。循環dna是存在于外周血中的有利于細胞外的核酸。目前檢測循環dna的方法有sanger一代測序和高通量測序及數字pcr等,sanger一代測序敏感性不到百分之十,高通量測序及數字pcr等敏感性最大可達到千分之一,但可能存在假陽性且現有千分之一的敏感性仍不足以對腫瘤進行早期診斷。
技術實現要素:
本發明所要解決的問題在于提供一種利用自殺報告基因選擇性富集突變核酸片段及其方法,通過選擇性破壞抑制含有一種特定基因型片段相關載體的擴增而不會抑制另外基因型片斷相關載體的擴增,達到將特定基因型片段進行富集的目的。
本發明的第一方面提供的技術方案:以自殺基因作為報告基因選擇性富集核酸樣本中的突變核酸片段,以選擇性抑制核酸樣本中含等位基因的野生型基因片段的擴增,而不抑制核酸樣本中含等位基因的突變型基因片段的擴增。從而為測序提供一種已經對突變基因進行了富集的待測標本,以提高測序分析的敏感性。
本發明優選的技術方案中,以含有自殺基因的質粒載體選擇性富集核酸樣本中的突變核酸片段,優選以含自殺基因pdoner211質粒載體選擇性富集核酸樣本中的突變核酸片段。且自殺基因的序列為seqidno.1所示。
本發明優選的技術方案中,所述核酸樣本中含等位基因的野生型基因片段不含有終止密碼子,而突變型基因片段含有終止密碼突變或含有可轉化為終止密碼子的突變,即突變型基因片段在克隆進自殺載體后始終表現出含有終止密碼子的作用而使自殺基因喪失自殺作用。
本發明優選的技術方案中,核酸樣本中含等位基因的野生型基因片段或突變型基因片段為10-200bp;更優選地,所述野生型基因片段或突變型基因片段為20-100bp;進一步,所述野生型基因片段或突變型基因片段優選為10-50bp。
本發明優選的技術方案中,所述核酸樣本中含等位基因的突變型基因片段為點突變或移碼突變。
優選地,核酸樣本中含等位基因的野生型基因片段為10-200bp,且不含終止密碼子;更優選地,所述野生型基因片段為20-100bp,且不含終止密碼子;進一步所述野生型基因片段優選為10-50bp,且不含終止密碼子。
本發明的第二方面提供自殺基因作為報告基因進行選擇性富集核酸樣本中的突變核酸片段,其特征在于,其包括如下步驟:
(1)評估核酸樣本情況,
(2)提供含有自殺基因作為報告基因的質粒載體,
(3)將步驟(1)中的核酸樣本連接到步驟(2)中的含有自殺基因作為報告基因的質粒載體中,
(4)將步驟(3)中的質粒載體轉化h5a感受態細胞,在含有抗性篩選的瓊脂培養基上培養富集,
(5)挑取篩選培養基上的菌落,測序驗證。
本發明優選的技術方案中,所述步驟(1)中核酸樣本中含等位基因的野生型基因片段或突變型基因片段為10-200bp,且不含終止密碼子時,評估適于本方法進行后續操作。
本發明優選的技術方案中,所述步驟(2)中為含有自殺基因的質粒載體,所述步驟(3)中將核酸樣本連接到含有自殺基因的質粒載體中;優選地為pdoner211質粒載體。
本發明優選的技術方案中,核酸樣本構建的兩側酶切位點可以包括但不限為:aari,hindiii或kpni酶切位點。優選地,步驟(3)中將核酸樣本連接到pdoner211質粒載體的多克隆位點中。
本發明優選的技術方案中,自殺基因作為報告基因進行選擇性富集核酸樣本中的突變核酸片段,其特征在于,其包括如下步驟:
(a)評估核酸樣本情況,
(b)提供含有自殺基因的質粒,
(c)以含有自殺基因的質粒為載體,將pcr擴增的待測標本片段克隆到含有自殺基因的的載體中,
(d)將步驟(c)中的質粒載體轉化h5a感受態細胞,在含有kana霉素抗性篩選的瓊脂培養基上培養富集,
(e)挑取篩選培養基上的菌落,pcr或者測序驗證。
優選地,所述核酸樣本中含等位基因的突變型基因片段的突變為點突變;若突變前,不含有終止密碼子,便不能正常在宿主細胞上生長;突變后,含有終止密碼子,能在宿主細胞上生長;優選為kras12位點突變,其突變之后含有終止密碼子,將這段序列克隆到自殺基因中,看他在自殺基因上的生長情況。
優選地,所述核酸樣本中含等位基因的突變型基因片段的突變為移碼突變;具體而言當突變核酸片段為3的整數倍3n時,由于片段的堿基數目的是3的整數倍,不產生移碼,含有該型質粒的菌落無法在培養基上生長;當突變核酸的片段不是3的整數倍,如為3n+1或者含有終止密碼子,含有該型質粒的菌落就可以在培養基上生長。
大腸桿菌db3.1細胞含有gyra462基因,對λ嗜菌體的ccdb基因產物的毒性具有抵抗作用,特別適用于轉化和擴增包含ccdb基因的質粒載體。使用puc19質粒檢測,轉化效率可達107cfu/μgdna.,-70℃保存幾個月轉化效率不發生改變。每支感受態可以酌情分裝使用,降低了實驗的成本。質量穩定,使用方便,質優價廉。
db3.13菌株的基因型為:f-gyra462enda1δ(sr1-reca)mcrbmrrhsds20(rb-,mb-)supe44ara-14galk2lacy1proa2rpsl20(smr)xyl-5λ-leumtl1抗生素耐藥性:db3.13感受態細胞具有鏈霉素抗性。操作步驟(以下操作均按無菌條件的標準進行):提示db3.1感受態細胞應保存在-70℃,不可多次凍融和放置時間過長,以避免降低感受態細胞的轉化效率。進行轉化操作時,應根據相應溫度及無菌條件的要求進行。為防止轉化實驗不成功,可以保留部分連接反應液,以重新轉化,將損失降到最低。
本發明中,自殺基因被選作為報告基因,因為它同時滿足了上述用于檢測基因突變的報告基因的兩個要求,對突變有極高耐受性,假陽性極少。如果插入片段的堿基數目的是三的整數倍,不產生移碼,該型質粒就不能在普通的感受態細胞上生長。如果插入的片段不是三的整數倍或者含有終止密碼子,該型質粒就可以在普通的大腸桿菌感受態細胞生長。通過長菌數目的比例,從而判斷出是野生還是突變,最后應用于kras熱點突變的12位突變點,突變之前,沒有終止密碼子。突變后,產生終止密碼子,從而可以在普通的大腸桿菌上面生長。最后按不同比例混合野生和突變的oligo,在野生與突變比例為10000:1的情況下,連接載體進行轉化,挑選2個生長的菌去測序,最后測序全部是突變。該方法提供了一種新型的檢測基因突變的方法。敏感性可達到萬分之一。
本發明中自殺基因被選作為報告基因,它同時滿足了兩個要求。對突變有極高耐受性,假陽性極少。試驗證明自殺基因在100bp以下(插入片段51bp,81bp)可以表現出理想模式,無假陽性產生。它可以作為用于檢測腫瘤原癌基因突變的報告基因。
通過選擇性破壞抑制含有一種特定基因型片段相關載體的擴增而不會抑制另外基因型片斷相關載體的擴增,達到將特定基因型片段進行富集的目的。本發明通過對標本中不同核酸基因型的富集構成,改變待測標本中不同基因型基因片段的比例,可用于基因突變分析。
本發明采用了一個新的方法來鑒定突變:運用寡核苷酸串來檢測目的基因的突變性。寡核苷酸串防止了突變的無意義性,并且使得插入基因片段的延伸和切斷具有了靈活性。在試驗中,發明人采用了自殺基因作為報告基因,它被證明同時具有兩個優點:可高度耐受突變,幾乎沒有假陽性。同時因為富集的待測標本是用于測序分析,本發明為提高測序分析的敏感性具有實用價值。
附圖說明
下面結合附圖及實施例對本發明作進一步描述:
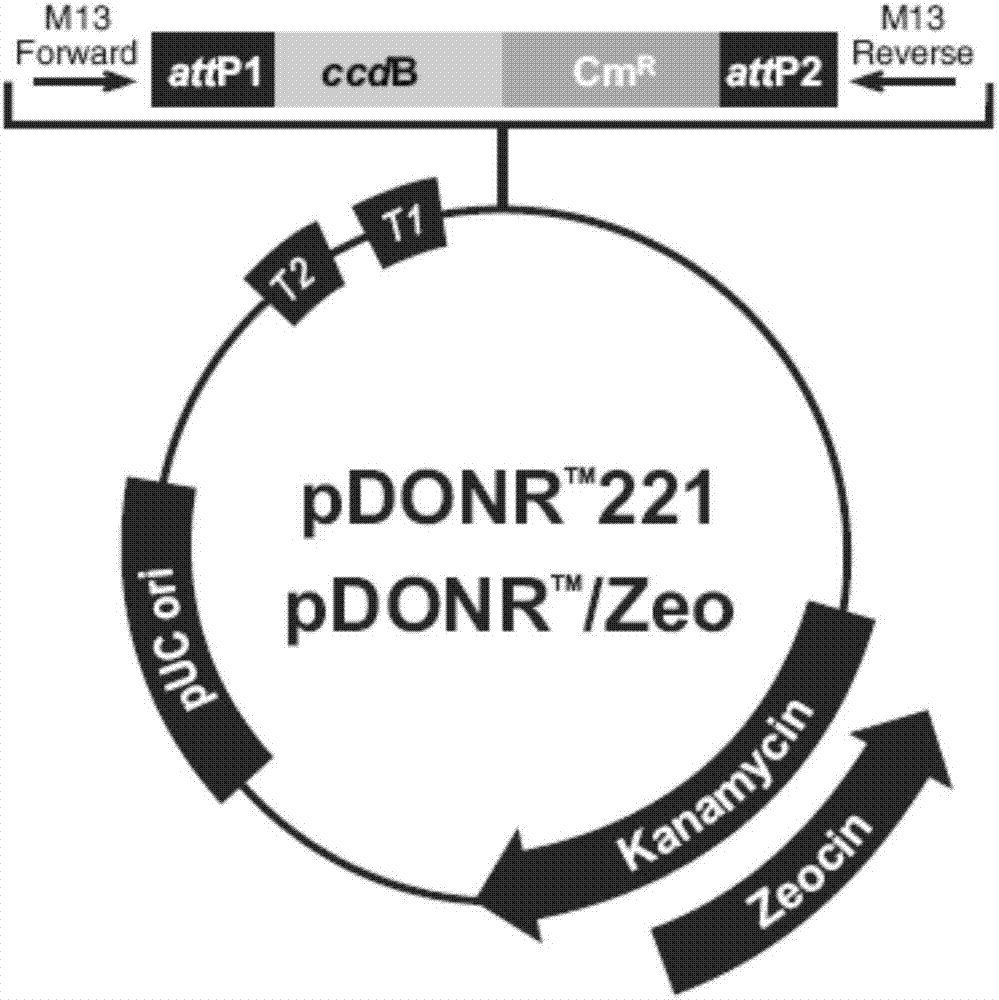
圖1為pdoner211的質粒圖譜。
圖2為重疊延伸加入aari酶切位點和hindiii位點說明圖。
圖3為在dh5a培養基上,移碼和不移碼的oligo片段菌落生長圖,其中,圖3a為3n菌落生長圖;圖3b為3n+1菌落生長圖;3n+1比3n長的多,3n并不是完全不長菌,說明自殺基因的效率并不是100%的。
圖4在db3.1培養基上,移碼和不移碼的oligo片段菌落生長圖,其中,圖4a為3n菌落生長圖,圖4b為3n+1菌落生長圖。
圖5為插入報告基因的堿基為3n+1的測序圖。
圖6為插入報告基因的堿基為3n的測序圖。
具體實施方式
為了便于理解,以下將通過具體的實施例對本發明進行詳細地描述。需要特別指出的是,這些描述僅僅是示例性的描述,并不構成對本發明范圍的限制。依據本說明書的論述,本發明的許多變化、改變對所屬領域技術人員來說都是顯而易見的。
一.儀器、材料
引物合成及測序委托蘇州金唯智公司完成。pdoner211質粒購自invitrogen公司,其質粒圖譜如圖1所示,其上具有卡那霉素抗性基因。pcr擴增儀(abi公司,system9700),電泳儀(bio-rad生物公司),pcr擴增試劑盒,aari購于(購于newenglandbiolabs公司),hindiii和kpni相關限制性內切酶(購于thermoscientific公司),dna核酸片段回收試劑盒(購于天根生化)。t4dna連接酶(購于thermoscientific公司),其余均為國產試劑及耗材。
二.建立方法
實施例1
提供pdoner211質粒,并以pdoner211質粒為模板引入aari的酶切位點
1.以pdoner211質粒為模板,pcr重疊延伸片段引入aari的酶切位點,
1.1用pdoner211質粒作為模板,利用引物xi+(seqidno.2)和引物xi-(seqidno.3)進行pcr擴增,每20μlpcr反應體系包括:2.0μl的10xpfubufferwithmgso4,0.3μl的pfupolymerase(2.5u/μl),0.5μl的dntpmix(10mmeach),兩條特異性引物分別為0.5μl,模板0.5μl,余量為水。pcr反應條件為:95℃預變性5min;95℃變性30s,58℃退火30s,72℃延伸30s,共30個循環;最后72℃5min。
得到的產物目的片段編號為片段1和片段2。
1.2用pdoner211作為模板,利用引物hi+(seqidno.4)和hi-(seqidno.5)進行pcr擴增,每20μlpcr反應體系包括:2.0μl的10xpfubufferwithmgso4,0.3μl的pfupolymerase(2.5u/μl),0.5μl的dntpmix(10mmeach),兩條特異性引物分別為0.5μl,模板0.5μl,余量為水。pcr反應條件為:95℃預變性5min;95℃變性30s,58℃退火30s,72℃延伸30s,共30個循環;最后72℃5min。
得到的產物目的片段編號為片段3和片段4。
1.3將上述1.1得到產物目的片段1和片段2,將上述1.2得到產物目的片段3和片段4,在37攝氏度環境下放置30-60分鐘。
1.4以上述步驟1.3孵育的產物作為新的模板,用primerxi+和primerhi-做pcr擴增,每20μlpcr反應體系包括:1.0μl的10xpfubufferwithmgso4,0.3μl的pfupolymerase(2.5u/μl),0.5μl的dntpmix(10mmeach),xi+引物為0.5μl,xi-引物為0.5μl,目的片段1、2、3、4合計為10.0μl,水為7.2。pcr反應條件為:95℃預變性5min;95℃變性30s,58℃退火30s,72℃延伸30s,共30個循環;最后72℃5min。
所獲得的目標產物記作1-4,即為攜帶arri酶切位點核酸片段,酶切后,則可以用來插入攜帶自殺基因的plasmid。
2.將前述含有aari酶切位點的片段,連接到pdoner211質粒中
2.1用bamhi和psti酶切pdoner211質粒和步驟1.4得到的pcr產物,酶切體系反應條件為37℃孵育60分鐘。然后用tiangen通用型dna純化回收試劑盒回收酶切片段,
2.2將上述兩個酶切產物進行連接反應,連接體系如下:t4buffer0.5μl,pcr產物3.0μl,pdoner211質粒1.0μl,t4酶1μl,最后補充水到10μl。反應條件為:22°環境下孵育1小時,產物即為帶有kana霉素抗性基因和自殺基因以及aar1酶切位點的plasmidpdonr211的連接混合液。
aari酶切位點的優勢:保證無縫酶切,不殘留酶切位點。
2.3取上述的連接混合液,轉化h5a感受態細胞,然后涂布kana篩選培養基,具體方法為:取50μl的dh5a感受態細胞在冰上融化10分鐘,并且加入上一步驟的連接體系,隨后放置冰塊上三十分鐘,42℃水浴熱擊42秒,快速置于冰上15分鐘,隨后加入800μllb培養基,(無抗生素),180轉,37℃振蕩培養40分鐘。離心后分離棄上清液(500微升),剩下的300微升吹打均勻,涂在含有kana霉素的瓊脂培養基上,放置在37℃的恒溫培養箱內培養一夜,第二天觀察菌落是否有形成。
還可以隨機挑取上述菌落進行pcr或者測序檢測,進行進一步的確認。
實施例二
在實施1中制備得到的含有aari酶切位點的pdoner211質粒中插入寡核苷酸串(移碼突變)
如果插入片段的堿基數目的是三的整數倍,則不產生移碼,含有該型質粒的菌落無法在培養基上生長;如果插入的片段不是三的整數倍3n+1或者含有終止密碼子,含有該型質粒的菌落就可以在在培養基上生長。
3.1設計并合成oligo如下:
nnc3n+:aatgnncnncnncnncnncnncnncnnc
nnc3n-:actggnngnngnngnngnngnngnngnn
其中,nnc控制了終止密碼子不會出現。具體實例,nnc3n+(seqidno.6),nnc3n-
(seqidno.7)。
3.2pdoner211質粒使用aar1進行單酶切,酶切體系為:10ⅹaar1buffer80μl,50ⅹoligo16μl,dna150μl,加水到體系為800μl。酶切完成后用tiangen回收,具體同前步驟。
3.3oligo退火及連接:將寡核苷酸串oligo的3n+和3n-退火,形成雙鏈dna,構建annealing體系,然后將產生的annealing產物稀釋十倍,再與載體pdoner211混合,連接體系如下:t4actbuffer1μl,t4酶1μl,回收的pdnoer2111μl,退火片段3n1μl,加水到體系為10μl;16℃過夜連接。
3.4將上述的連接產物分別轉化感受態細胞dh5a和db3.1,涂在含有kana霉素的瓊脂培養基上,放置在37℃的恒溫培養箱內培養過夜,第二天觀察兩種感受態細胞形成菌落的情況。次日直接挑菌測序。
3.5設計并合成oligo如下:
nnc3n+1+:aatgnncnncnncnncnncnncnncnncn
nnc3n+1-:actgngnngnngnngnngnngnngnngnn
具體實例,可以為:nnc3n+1+(seqidno.8)和nnc3n+1-(seqidno.9)。
3.6pdoner211質粒使用aar1進行單酶切,酶切體系為:10ⅹaar1buffer80μl,50ⅹoligo16μl,dna150μl,加水到體系為800μl。酶切完成后用tiangen回收,具體同前步驟。
3.7oligo退火及連接:將寡核苷酸串oligo的3n+1+和3n+1-退火,形成雙鏈dna,構建annealing體系,然后將產生的annealing產物稀釋十倍,再與載體pdoner211混合,連接體系如下:t4actbuffer1μl,t4酶1μl,回收的pdnoer2111μl,退火片段3n1μl,加水到體系為10μl;16℃過夜連接。
3.8將上述的連接產物分別轉化感受態細胞dh5a和db3.1,涂在含有kana霉素的瓊脂培養基上,放置在37℃的恒溫培養箱內培養過夜,第二天觀察兩種感受態細胞形成菌落的情況。次日直接挑菌測序。
實施例三將kras12基因連接到實施例1制備得到的含有aari酶切位點的pdoner211質粒中,進行選擇性富集kras12基因突變核酸片段
1.更換aari酶切位點為kpni和hindiii,
備注:hindiii:aagctt,kpni:ggtacc
設計oligo,其中oligo+(seqidno.10)和oligo-(seqidno.11)。
a.aari酶切新構建好的質粒
b.annealoligo+,oligo-
c.連接酶切好的載體和oligo,轉化。
2.雙酶切,加入基因組kras序列
設計如下片段oligokt50+(seqidno.12),oligokt50-(seqidno.13),oligokw50+(seqidno.14),oligokw50-(seqidno.15)。
hindiii,kpni雙酶切上述構建好的質粒。anneal:oligokt50+,oligokt50-;oligokw50+,oligokw50-。
然后連接雙酶切的載體和oligo。
3.連接轉化,及進行挑菌檢測
kras野生突變10:1,100:1,1000:1,10000:1不同比例混合,然后挑菌搖菌提取dna,送測序,看里面是否有突變的菌。即在萬分之一的突變長菌培養皿中,挑的兩個菌去測序,測序全部正確,符合預期。
三、結果分析
理論上來講,攜帶了自殺基因的片段導入dh5a后,其表達的酶可催化產生細胞毒物質,從而殺死攜帶該基因的受體細胞即dh5a,所以菌落無法在培養基上生長。當基因片段移碼(堿基數目變為3n+1)過后,這種酶無法再分泌,受體細胞也就能夠存活,菌落正常生長。
1.在突變比例為萬分之一比例的情況下,通過自殺基因選擇性富集突變型的基因,抑制野生型的基因,最后在轉化鋪板的過程中,通過挑菌搖菌提取dna,能夠測序到突變的菌,提供了一種新型的檢測突變型的方法。kras12位點突變,突變之后含有終止密碼子,將這段序列克隆到自殺基因中,看他在自殺基因上的生長情況。若突變前,不含有終止密碼子,便不能正常在宿主細胞上生長;突變后,含有終止密碼子,能在宿主細胞上生長。
2.dh5a生長情況
在dh5a培養基上,移碼和不移碼的oligo片段以同樣的plasmid濃度(20n克)轉化,長菌的比例為3n+1:3n=2000:1。
3.db3.1生長情況
在db3.1培養基上,不管移碼還是不移碼的plasmid,都長菌落,db3.1是一種特異的感受態細胞。
4.pcr片段在dh5a上的生長
這一步是為了檢測只是單純移碼,不受終止密碼子的影響,plasmid是否能轉化生長,驗結果是可以的。
將兩種寡核苷酸串:3n+1和3n,插入pdoner211質粒中,如果插入片段的堿基數目的是三的整數倍,不產生移碼,該型質粒就只能在db3.1的感受態細胞生長;如果插入的片段不是三的整數倍或者含有終止密碼子,該型質粒就可以在普通的大腸桿菌感受態細胞生長,
上述實例只為說明本發明的技術構思及特點,其目的在于讓熟悉此項技術的人是能夠了解本發明的內容并據以實施,并不能以此限制本發明的保護范圍。凡根據本發明精神實質所做的等效變換或修飾,都應涵蓋在本發明的保護范圍之內。
序列表
<110>蘇州佳章生物技術有限公司
<120>利用報告基因選擇性富集突變核酸片段及其方法
<160>15
<210>1
<211>255
<212>dna
<213>合成
<400>1
atgcagtttaaggtttacacctataaaagagagagccgttatcgtctgtttgtggatgta60
cagagtgatattattgacacgcccgggcgacggatggtgatccccctggccagtgcacgt120
ctgctgtcagataaagtctcccgtgaactttacccggtggtgcatatcggggatgaaagc180
tggcgcatgatgaccaccgatatggccagtgtgccggtctccgttatcggggaagaagtg240
gctgatctcagccac255
<210>2
<211>28
<212>dna
<213>合成
<400>2
cgcgtggatccggcttactaaaagccag28
<210>3
<211>50
<212>dna
<213>合成
<400>3
cgatgcaggtgaagcttcacctgccgatcatttcaccagcccctgttctc50
<210>4
<211>50
<212>dna
<213>合成
<400>4
gtgaagcttcacctgcatcgcagtttaaggtttacacctataaaagagag50
<210>5
<211>25
<212>dna
<213>合成
<400>5
actggccatatcggtggtcatcatg25
<210>6
<211>28
<212>dna
<213>合成
<400>6
aatgccctccctccccttctccgtcttc28
<210>7
<211>28
<212>dna
<213>合成
<400>7
actggaagacggagaaggggagggaggg28
<210>8
<211>29
<212>dna
<213>合成
<400>8
aatggacgcccccctcgcctacaccgccc29
<210>9
<211>29
<212>dna
<213>合成
<400>9
actggggcggtgtaggcgaggggggcgtc29
<210>10
<211>23
<212>dna
<213>合成
<400>10
aatgaagcttnncnnccggtacc23
<210>11
<211>23
<212>dna
<213>合成
<400>11
actgggtaccggnngnnaagctt23
<210>12
<211>61
<212>dna
<213>合成
<400>12
agcttacttgtggtagttggagctgatggcgtaggcaagagtgccttgacgatacaggta60
c61
<210>13
<211>53
<212>dna
<213>合成
<400>13
ctgtatcgtcaaggcactcttgcctacgccatcagctccaactaccacaagta53
<210>14
<211>61
<212>dna
<213>合成
<400>14
agcttacttgtggtagttggagctggtggcgtaggcaagagtgccttgacgatacaggta60
c61
<210>15
<211>53
<212>dna
<213>合成
<400>15
ctgtatcgtcaaggcactcttgcctacgccaccagctccaactaccacaagta53
- 還沒有人留言評論。精彩留言會獲得點贊!