桑葚病原菌高通量鑒定及種屬分類方法及其應用與流程
本發明涉及植物病原菌鑒定及種屬分類技術領域,更具體地,涉及一種桑葚病原菌高通量鑒定及種屬分類的方法。
背景技術:
近年來,隨著蠶桑多元化發展和桑產業的興起,桑不僅是以桑葉作為蠶桑產業中特種經濟動物——蠶的飼料,更因其桑樹本身的藥理及營養價值,使得桑的多用途資源利用變得更加豐富。桑樹全身都是寶,桑葉、桑椹和桑枝是目前種桑的主要目的收獲物,桑椹作為桑科桑屬植物的果實,又稱桑果、桑葚或桑棗,被譽為“民間圣果”,自古以來就是百姓常采用的一種利尿,保健,消暑的鮮果,可制作果干、果汁,也可來泡酒。桑椹性味甘寒,具有補肝益腎、生津潤燥、烏發明目等功效。因此桑果是人們植桑時,因桑葚具有較高的營養及食用價值,故桑果是除收獲桑葉外具有較大經濟價值的桑產物(陳冬梅,2013)。
對桑果威脅最大的病害為桑菌核病。桑菌核病是果桑一種主要病害,俗稱桑白果病,屬真菌類病害;桑樹開花時病菌開始侵入,桑果快成熟時病狀顯現,顏色呈白色,失去經濟價值,大多數的果桑品種均較易發病,發病率可高達90%以上(蒯元璋等,2012)。
桑椹菌核病(popcorn disease)分桑椹肥大性菌核病、桑椹縮小性菌核病和桑椹小粒性菌核病3種,俗稱白果病。桑椹菌核病病菌以菌核在土壤中越冬,越冬后春季溫暖、多雨、土壤潮濕利于菌核萌發,產生子囊盤多,病害重。在通風透光差,低洼多濕的桑園更適合病原菌的萌發與生長;花果多,樹齡老的桑園,隨著果桑養成年份的增加,菌核病病原(菌核或子囊盤)的田間殘留量增加,也加大了菌核病爆發的可能性(婁利峰等,2016)。
桑椹肥大性菌核病的植株花被厚腫,顏色呈現灰白,果實比較膨大,果實的中心有個黑色菌核,得病的桑椹破碎后會聞到一股臭味;感染縮小性菌核病的果實縮小,明顯顏色呈現灰白,質地變得比較堅硬,表面會有暗褐色的細斑長出,內部存在黑硬菌核;小粒性菌核病的癥狀是果實體積膨大,果實內部長有小粒形菌核,果實呈現出病態的灰黑色,易脫落。
桑椹肥大性菌核病病菌Ciboria shiraiana.稱白杯盤菌(桑實杯盤菌),屬子囊菌門真菌。分生孢子梗叢生,基部粗頂端細小,上生分生孢子。分生孢子單胞,卵形,無色。菌核萌發產生1-5個子囊盤,盤內生子囊,側絲細長,內有8個子囊孢子,子囊孢子橢圓形,無色單胞,具隔膜1-2個。
桑椹縮小性菌核病病原菌為白井地杖菌Mitrula shiraiana,其分生孢子梗細絲狀,具分枝,端生,卵至橢圓形、無色。單胞型分生孢子菌核會以兩種方式長出子實體。子實體有灰褐色、扁平長柄,長有茸毛;子實體頭部長橢圓形,具數條淺褐色縱向排列的稻紋,子囊生在頭部外側子實層里,長棍棒狀,先端圓,基部細,內生8個子囊孢子,子囊孢子單胞、無色、橢圓形。
桑椹小粒性菌核病病原菌為肉阜狀杯盤菌Ciboria canrunculoides,分生孢子接近于球形,較圓,子囊盤杯狀,具長柄,子囊的形狀近似于長棍棒,內部含有8個子囊孢子,子囊孢子單胞,無色,腎形,有半球形小體附著。側絲有分枝,有隔或無隔。
桑菌核病致病菌除上述的桑實杯盤菌、白井地杖菌、肉阜狀杯盤菌外,最近陳倩茜(2015)報道從桑菌核病的樣本中分離確定了一種新的病原菌,鑒定為擬莖點霉屬(Phomopsis)的病原真菌Phomopsis sp.SC1104,該病原菌可能與桑椹菌核病流行爆發有關。桑菌核病致病菌的寄主多樣,病原可危害32科160多種植物,而桑菌核病還可與油菜等十字花科植物菌核病病原菌交叉感染(范錦等,2014;呂蕊花等,2015),這給桑菌核病的防治帶來了困難。
植物病原菌分類一般以寄主的名稱作為真菌種名的命名原則,同科同屬且存在于同一寄主上的真菌無法在命名上和形態上區分開來,而若同種病原菌在不同的寄主間出現交叉感染情況,該病原菌的命名變得復雜而難以鑒別或區分,類似桑葚菌核病有多種病原菌感染的情況,對于其主要感染病原菌的鑒別,則需要分子生物學的技術加以輔助鑒別。
真菌的系統分類方法中,更多的是以遺傳物質的信息來佐證傳統形態學的分類,并糾正一些主觀分類的錯誤。現有的研究方法中,常用核糖體DNA(ribosome DNA,rDNA)序列進行測序比對。rDNA中編碼18S、5.8S、28S的rDNA序列較為保守,可用于真菌目、科、屬等分類階元的系統發育研究;內轉錄間隔區ITS位于核糖體rDNA 18S、5.8S及28S之間,由于不需要加入成熟核糖體,因此在進化過程中能夠承受更多的變異,其進化速率為18S rDNA的10倍,屬于中度保守的區域,利用它可研究種及種以下的分類階元。
全基因組測序作為已經成熟且推廣開來的一種測序,多用于基因的功能注釋或進一步的比較基因組學研究。全基因組的高通量測序多用于已知菌株的功能基因的查找鑒定,用于真菌的種屬鑒定,其準確性毋庸置疑。基于分子水平判定物種的技術已較為成熟,而測序的快速與成本低廉也使得這項技術變得更大眾化。針對現有的桑葚菌核病病原菌的鑒定及種屬分類研究中,受傳統研究方法的限制,缺乏高通量技術、結果往往欠準確性,對桑葚菌核病病原菌進行定量檢測方面幾乎空白,因此本發明的方法具有前瞻性。
技術實現要素:
本發明要解決的技術問題是針對現有對桑葚病原菌鑒定不夠準確的技術不足,提供一種準確性更高同時種屬分類明確的鑒定及物種分類方法,并且檢測對象是基于桑葚。
本發明要解決的另一技術問題是提供所述方法在對桑葚病原菌進行定量分析方面的應用。
本發明還一要解決的技術問題是提供所述方法在分析微生物物種豐度方面的應用。
本發明還一要解決的技術問題是提供所述方法在計算物種間遺傳距離方面的應用。
本發明的目的通過以下技術方案予以實現:
提供一種桑葚病原菌高通量鑒定及種屬分類的方法,利用高通量測序手段及生物信息學分析方法,建立一種桑葚病原菌的快速、準確、高效的鑒定及種屬分類方法。
具體地,所述鑒定及種屬分類方法包括以下步驟:
S1.收集桑葚病果;
S2.提取桑葚病果的總DNA;
S3.Illumina DNA文庫構建;
S4.Illumina高通量測序;
S5.去除測序數據中桑樹基因組序列;
S6.組裝微生物基因組序列;
S7.組裝完整核糖體序列;
S8.篩選標記微生物核糖體DNA序列;
S9.對比分析核糖體DNA序列進行種屬分類;
其中,步驟S2所述提取桑葚病果的總DNA的方法為:剪取桑病果中的患病區域,加入液氮充分研磨,用真菌DNA提取試劑盒按照其操作說明提取病葉總DNA,所述總DNA保存于-20℃冰箱;
步驟S3所述Illumina DNA文庫構建的方法為:依照Illumina文庫構建流程,將所述步驟S2中所述總DNA構建為片段大小400-600bp范圍內的的雙末端高通量測序文庫;
步驟S4所述Illumina高通量測序的方法為:用Illumina Hiseq 2500測序儀對所述步驟S3中所述DNA文庫進行高通量測序。
步驟S5所述去除測序數據中桑樹基因組序列的方法為:利用比對軟件對步驟S4中所述高通量測序數據進行數據比對分析。選擇比對算法,將所述測序數據與桑樹參考基因組進行比對,并將比對上參考基因組的所述測序數據判定為桑樹基因組序列。使用編寫的計算機程序從所述測序數據中去除桑樹基因組序列。
S5所述去除桑樹的基因組DNA序列選用的參考基因組序列為:
桑屬川桑(Morus notabilis)全基因組序列(GCA_000414095.2)和葉綠體基因組序列(NC_027110.1)。
優選地,所述比對軟件為bwa(0.7.12-r1039)軟件;
優選地,所述比對算法為mem比對算法;
優選地,所述將測序數據與桑樹參考基因組進行比對選用的是雙末端比對方法和bwa(0.7.12-r1039)軟件的默認參數;
優選地,所述編寫的計算機程序是用python計算機語言編寫。
步驟S6所述組裝微生物基因組序列的方法為:使用組裝軟件對步驟S5所述已去除桑樹基因組序列的測序數據進行組裝。
優選地,所述組裝軟件為MetaVelvet(v1.2.01)。
步驟S7所述組裝完整核糖體DNA序列的方法為:采用比對軟件,對所述組裝序列進行比對,根據比對結果從測序數據中獲取雙末端測序片段,使用組裝軟件對序列進行組裝和延伸,經多個循環操作,直至獲得完整的核糖體DNA序列;
優選地,所述比對軟件為bwa(0.7.12-r1039)軟件;
優選地,所述比對方法采用無錯配(0mismatch)和無空缺(0gap)比對;
優選地,所述組裝軟件為MetaVelvet(v1.2.01)軟件。
步驟S8所述篩選標記微生物核糖體DNA序列的方法為:使用序列比對分析軟件,設定比對期望值,將步驟S6所述微生物基因組序列與數據庫進行比對,根據比對結果對序列標簽進行注釋及物種分析;
優選地,所述比對期望值<1e-20;
優選地,所述序列比對分析軟件為blastn(2.2.31+)軟件;
優選地,所述數據庫為NCBI數據庫中的nt數據庫。
步驟S9所述對比分析核糖體DNA序列進行種屬分類的方法為:使用軟件1對所述微生物核糖體DNA序列進行統計分析,選取系統樹構建模型,將所述模型代入軟件2中,構建系統進化樹。
優選地,所述軟件1為jModelTest2(https://github.com/ddarriba/jmodeltest2)在線軟件;
優選地,所述系統樹構建模型為取AIC(Akaike Information Criterion)值最小的替代模型GTR+G;
優選地,所述軟件2為RaxML(8.1.5);
優選地,所述構建系統進化樹的方法為使用最大似然法構建的系統進化樹,進化樹迭代次數為1000次。
本發明同時提供所述桑葚病原菌高通量鑒定及種屬分類方法在桑樹病原菌定量分析方面的應用。
本發明還提供所述桑葚病原菌高通量鑒定及種屬分類方法在分析微生物物種豐度方面的應用。
所述應用的方法為:使用分析軟件,計算核糖體DNA片段的平均測序深度,以此作為該物種的豐度值。
優選地,所述分析軟件為bwa(0.7.12-r1039)與samtools(v1.2)。
本發明還提供所述桑葚病原菌高通量鑒定及種屬分類方法在計算物種間遺傳距離方面的應用。
附圖說明
圖1桑葚菌核病病果所檢測到的真菌微生物分類樹。
圖2驗證核糖體組裝結果的兩對引物與兩對通用引物ITS1/ITS4、ITS4/ITS5電泳圖。
圖3核糖體DNA序列組成與堿基GC比例分布圖。
圖4菌核病病原體Ciboria carunculoides 18S進化樹。
圖5菌核病病原體Ciboria carunculoides ITS進化樹。
圖6菌核病病原體Ciboria carunculoides28S進化樹。
圖7菌核病病原體rRNA全基因序列進化樹。
具體實施方式
下面結合具體實施例進一步說明本發明方法。下述實施例和附圖僅用于示例性說明,不能理解為對本發明的限制。除非特別說明,下述實施例中使用的試劑原料為常規市購或商業途徑獲得的生試劑原料,除非特別說明,下述實施例中使用的方法和設備為本領域常規使用的方法和設備
本發明以對桑葚菌核病病原菌的鑒定及種屬分類為例,根據本發明思想,本領域技術人員可以參照現有技術,對其他病癥的病原菌進行鑒定及種屬分類。
實施例1
在發病的桑園尋找到具有典型桑葚菌核病病征的桑果,收集,剪取桑病果中的患病區域,剪下的材料使用液氮充分研磨,總DNA的提取使用康為世紀公司的真菌DNA提取試劑盒,具體按照其操作說明進行,提取后的總DNA保存于-20℃。依照Illumina文庫構建流程,將總DNA構建為片段大小450bp的雙末端高通量測序文庫,使用Illumina Hiseq2500測序儀對構建好的DNA文庫進行高通量測序,共測得9.03M對測序片段,測序讀長為雙末端125bp,總測序數據量2.26Gb。
由于DNA提取過程中包含桑樹的桑果材料,為減少測序數據中桑樹基因組數據對微生物序列組裝的影響,在進行微生物序列組裝前首先去除桑樹的基因組DNA序列。選取桑樹Morus notabilis全基因組序列(GCA_000414095.2)和葉綠體基因組序列(NC_027110.1)作為參考基因組序列,采用bwa(0.7.12-r1039)比對軟件進行數據比對分析。比對選擇mem比對算法,使用雙末端比對方法和軟件的默認參數,將測序數據與桑樹參考基因組進行比對,并將比對上參考基因組的測序片段判定為桑樹基因組序列。使用python編寫的計算機程序從fastq測序數據中去除桑樹測序數據,然后再進入微生物序列組裝。微生物序列的組裝使用MetaVelvet(v1.2.01)組裝軟件進行。
序列標簽注釋使用blastn(2.2.31+)序列比對分析軟件,組裝的序列標簽序列與NCBI的nt數據庫進行比對,blastn比對設定期望值<1e-20,根據比對結果對序列標簽進行注釋。核糖體DNA序列是細菌和真菌鑒定的重要的最常用的分子標記,因此物種分類鑒定和定量以核糖體DNA為主要分子標記。根據序列標簽注釋結果,選擇核糖體DNA序列作為微生物鑒定與定量分析依據。使用bwa(0.7.12-r1039)+samtools(v1.2)分析軟件,計算測序數據中核糖體DNA片段的平均測序深度,并以此作為該物種的豐度值。
結果顯示,共注釋228條rRNA序列標簽,數據包含4條細菌序列標簽,82條真菌序列標簽,分別對應3種細菌,3種真菌。通過對序列標簽注釋結果的查詢,共發現桑病果內存在3個屬的真菌微生物,相對豐度最高的為Ciboria屬的真菌,見附圖1,其分子標簽測序深度為1115,相對豐度為98.50,所占比例最高,相關文獻報道該屬絕大部分為植物病原,大部分導致植物出現果實或種子僵化膨大等病征,與本發明研究的病征相符合。
真菌的核糖體DNA由18S區段,ITS1區段,5.8S區段,ITS2區段和28S區段組成,序列總長度約為5.5Kb。MetaVelvet(v1.2.01)初始組裝的序列標簽為斷裂的核糖體標簽,為得到完整的核糖體DNA序列,分析采用序列捕獲和從頭組裝策略,以組裝完整的核糖體DNA。選擇包含目標病原菌ITS序列的核糖體DNA序列為參考序列,采用bwa(0.7.12-r1039)軟件進行0mismatch和0gap比對,根據比對結果從測序數據中獲取雙末端測序片段,進一步采用MetaVelvet(v1.2.01)組裝軟件對序列進行組裝和延伸,經多個循環操作,獲取完整的核糖體DNA序列。
根據組裝完整的核糖體DNA序列,采用真菌通用引物ITS1和ITS4、ITS4和ITS5(White,1990),和基于本研究組裝獲得的完整核糖體DNA序列設計了PCR驗證引物見表1,然后進行PCR擴增和Sanger測序方法,驗證組裝的正確性。通用引物序列見表2所示,PCR反應體系見表3所示。
表1.PCR驗證引物序列表
表2真菌ITS區域通用引物序列
表3 PCR反應體系(20μL)
真菌通用引物ITS1和ITS4引物組PCR程序:預變性94℃5min;94℃30s,55℃30s,72℃1min,30個循環;72℃10min。
真菌通用引物ITS4和ITS5引物組PCR程序:預變性94℃5min;94℃30s,55℃30s,72℃1min,30個循環;72℃10min。
驗證核糖體組裝結果的P1-F/R引物組PCR反應體系(20μL)PCR程序:94℃5min;94℃1min,56℃1min,72℃4min,30個循環;72℃10min。
驗證核糖體組裝結果的P2-F/R引物組PCR反應體系(20μL)PCR程序:94℃5min;94℃1min,56℃1min,72℃2min30s,30個循環;72℃10min。
取3μLPCR擴增產物用1.2%的瓊脂糖凝膠(EB染色)電泳檢測。通過瓊脂糖凝膠電泳回收大小對應的PCR產物片段,見附圖1所示,圖1中,M:Takara DL5000Marker;1.真菌通用引物ITS1和ITS4;2.真菌通用引物ITS4和ITS5;3.驗證核糖體組裝結果引物P1-F和P1-R;4.驗證核糖體組裝結果引物P2-F和P2-R。
PCR驗證結果見附圖2,從圖中可以看出,本次樣品較純凈,泳道1和2的結果說明用真菌通用引物ITS1和ITS4、ITS4和ITS5擴增的模板DNA是屬于真菌的DNA;泳道3和4的結果說明驗證核糖體組裝結果的P1引物組和P2引物組均擴增到相應的目的條帶,進一步測序后的結果與組裝的結果高度一致。
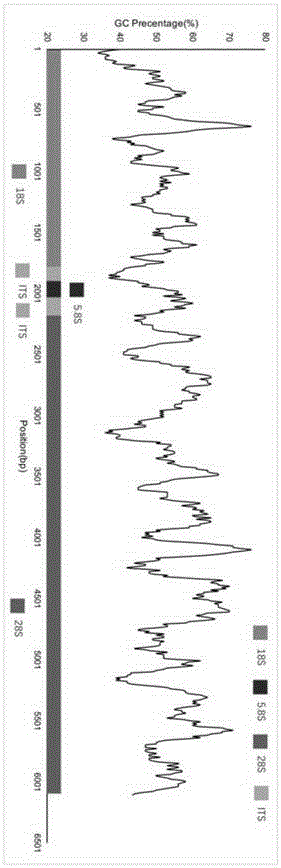
結果顯示,根據高通量測序數據組裝與實驗驗證結果,組裝得到完整核糖體DNA序列,見附圖3,長度6202bp(GC比例52.98%)。序列包含18S區域,ITS1區域,5.8S區域,ITS2區域,28S區域。其中18S區域長度1782bp(GC比例50.05%),ITS1區域長度101bp(GC比例43.56%),5.8S區域長度151bp(GC比例46.36%),ITS2區域長161bp(GC比例56.52%),28S區域長4007bp(GC比例54.63%)。18S區域和28S區域長度較大,占序列總長度的96.37%;而在GC比例上,ITS2區域(56.52%)的GC比例則明顯高于其他區域。
構建了對應Ciboria carunculoides的rRNA分子系統樹模型,菌核病病原體Ciboria carunculoides 18S進化樹、菌核病病原體Ciboria carunculoides ITS進化樹、菌核病病原體Ciboria carunculoides 28S進化樹、菌核病病原體Ciboria carunculoides的rRNA全基因序列進化樹分別見附圖4~附圖7所示。
使用blastn(2.2.31+)比對軟件將核糖體DNA與nt數據庫進行比對,對核糖體DNA序列進行注釋,共注釋出28S區域,ITS1區域,5.8S區域,ITS2區域和28S區域。使用python語言編寫的計算機程序對序列的GC比例進行分析,計算各區域的平均GC比例。同時程序以100bp為計算窗口,以10bp為步進,計算核糖體DNA的GC比例特征。
SEQUENCE LISTING
<110> 華南農業大學
<120> 桑葚病原菌高通量鑒定及種屬分類方法及其應用
<130>
<160> 7
<170> PatentIn version 3.3
<210> 1
<211> 20
<212> DNA
<213> 引物P1-F
<400> 1
taaataccga tgcggggctc 20
<210> 2
<211> 20
<212> DNA
<213> 引物P1-R
<400> 2
cactaggtcg ggacctctca 20
<210> 3
<211> 20
<212> DNA
<213> 引物P2-F
<400> 3
cgcttagcgt gctacccata 20
<210> 4
<211> 20
<212> DNA
<213> 引物P2-R
<400> 4
acaggaccgt gtaacgcaat 20
<210> 5
<211> 19
<212> DNA
<213> 通用引物ITS1
<400> 5
tccgtaggtg aacctgcgg 19
<210> 6
<211> 20
<212> DNA
<213> 通用引物ITS4
<400> 6
tcctccgctt attgatatgc 20
<210> 7
<211> 22
<212> DNA
<213> 通用引物ITS5
<400> 7
ggaagtaaaa gtcgtaacaa gg 22
- 還沒有人留言評論。精彩留言會獲得點贊!
- 一種構建Hi?C高通量測序文庫的方法與制造工藝
- 桑葚病原菌高通量鑒定及種屬分類方法及其應用與制造工藝
- 基于高通量測序技術檢測乳腺癌/卵巢癌易感基因的多重PCR特異性引物、試劑盒及方法與制造工藝
- 基于高通量測序檢測人EGFR基因變異的質控方法及試劑盒與制造工藝
- 基于高通量測序技術的小麥病原微生物檢測方法及其應用與制造工藝
- 一種基于二代高通量測序技術的地中海貧血癥的基因檢測試劑盒的制造方法與工藝
- 一種基于集群的高通量數據分析方法與制造工藝
- 高通量雙折射干涉成像光譜儀裝置及其成像方法與制造工藝
- 高通量測序檢測人循環腫瘤DNA EGFR基因的捕獲探針及試劑盒的制造方法與工藝
- 一種高通量測序檢測無創親子鑒定的方法與制造工藝