一種改性鎂鋁類水滑石/聚磷酸銨/成炭發泡劑/磷腈復配阻燃劑的制作方法
本發明涉及一種混合型膨脹阻燃劑。
背景技術:
隨著科學技術的發展,高分子材料的作用越來越重了,現在人們的生活中隨處可見由高分子材料制成的產品。阻燃劑的產生為高分子材料的擴大使用提供了重大的助力。
膨脹阻燃劑(ifr)屬于磷氮協同體系,主要由酸源(脫水劑),炭源(成炭劑)和氣源(發泡劑)組成,具有燃燒時煙霧少、放出氣體無害的特點。提高阻燃效率,降低阻燃劑的添加量是各類阻燃體系永恒的主題。
類水滑石作為新型阻燃劑,具有較好的發展空間,現如今對類水滑石阻燃劑的開發和應用主要集中在提高其與聚合物的相容性,但目前效果還不是太理想,急需開展進一步的研究和開發。
技術實現要素:
本發明的目的是提供一種改性鎂鋁類水滑石/聚磷酸銨/成炭發泡劑/磷腈復配阻燃劑,以硝酸鎂、硝酸鋁和混合堿為原料合成鎂鋁類水滑石,然后采用硬脂酸鈉對鎂鋁類水滑石進行表面改性,將表面改性后的鎂鋁類水滑石(ldh)與聚磷酸銨(app)、成炭發泡劑(cfa)和磷腈復配成具有協同作用的膨脹阻燃劑。
本發明的目的是通過以下技術方案實現的:
一種改性鎂鋁類水滑石/聚磷酸銨/成炭發泡劑/磷腈復配阻燃劑,由改性鎂鋁類水滑石(ldh)、聚磷酸銨(app)、成炭發泡劑(cfa)和磷腈按照1.6~3.2:24:8:0.8~2.4的質量比復配而成。
本發明中,采用共沉淀法和微波晶化法制備改性鎂鋁類水滑石,具體步驟如下:
(1)分別配置0.5~1.5mol/l的硝酸鎂、硝酸鋁的水溶液1l。
(2)取70ml硝酸鋁及280ml硝酸鎂得到混合鹽溶液;取500~600ml、0.6~1.2mnaoh和0.45~0.9mna2co3混合堿待用。
(3)在60~80℃水浴及不斷攪拌下,通過兩端的恒壓滴液漏斗向四口瓶中滴加混合鹽及混合堿,控制溶液ph為8~9,滴完后共沉淀反應0.5~1.5h。
(4)向溶液中加入0.2~0.25g硬脂酸鈉改性1~3h。
(5)將溶液倒入500ml燒杯中,并在30~90℃下微波晶化10~90分鐘。
(6)用蒸餾水將溶液洗滌至ph=7,抽濾、干燥,得到改性鎂鋁類水滑石。
本發明中,所述膦腈可以是所有類型的膦腈,比如:六氯環三膦腈、六對羧基苯氧基環三磷腈、苯氧基磷腈、對羧基苯氧基磷腈、聚氨基磷腈、烴氧基磷腈,六對羧基苯氧基環三磷腈的結構式如下:
本發明中,所述成炭發泡劑的化學結構式如下:
將本發明的復配阻燃劑添加到聚烯烴乙烯醋酸乙烯共聚物(eva)中(質量百分含量為1~60%),氧指數的測試結果表明:與只有類水滑石作為阻燃劑相比,復配阻燃劑的添加使復合材料的阻燃性能有明顯的提高。熱性能測試結果表明:阻燃劑的添加使得復合材料的熱失重速率溫度較純eva有明顯提高。力學性能測試結果表明:隨著改性鎂鋁類水滑石在復配阻燃劑中所占量的增加,其斷裂伸長率和拉伸強度值呈不規則下降。但是均比單獨添加磷腈阻燃劑和類水滑石化合物復合材料的力學性能有所提高。
將本發明的復配阻燃劑添加到聚丙烯(pp)中(質量百分含量為1~60%),氧指數和垂直燃燒的測試結果表明:與單獨類水滑石作為阻燃劑相比,復配阻燃劑的加入對聚丙烯的阻燃性能提高的更加明顯。力學性能測試結果指出隨著鎂鋁改性類水滑石在復配阻燃劑中所占比例的增加,其拉伸強度和斷裂伸長率值呈不規則下降,但均比單獨添加改性類水滑石的力學性能有所提高。
附圖說明
圖1為具體實施方式一中改性鎂鋁類水滑石(mgal-ldh)的xrd譜圖;
圖2為具體實施方式一中改性鎂鋁類水滑石(mgal-ldh)的ir譜圖;
圖3為具體實施方式一中ldh、app、cfa和磷腈的tg-dtg曲線,(a):ldh,(b):app,(c):cfa,(d):磷腈;
圖4為具體實施方式一中純eva和含有20wt.%含量阻燃劑的復合材料的tga曲線;
圖5為具體實施方式一中純eva和含有20wt.%含量阻燃劑的復合材料的dtg曲線,(a):eva180,(b):eva144ldh36,(c)eva144app36,(d)eva144cfa36,(e)eva144磷腈36,(f)eva144ldh3.2app24cfa8磷腈0.8,(g)eva144ldh3.2app24cfa8磷腈0.8,(h)eva144ldh3.2app24cfa8磷腈0.8;
圖6為具體實施方式一中純eva和復合材料樣品的氧指數柱形圖;
圖7為具體實施方式一中純eva和復合材料樣品的拉伸強度柱狀圖;
圖8為具體實施方式一中純eva和復合材料樣品的斷裂伸長率柱狀圖;
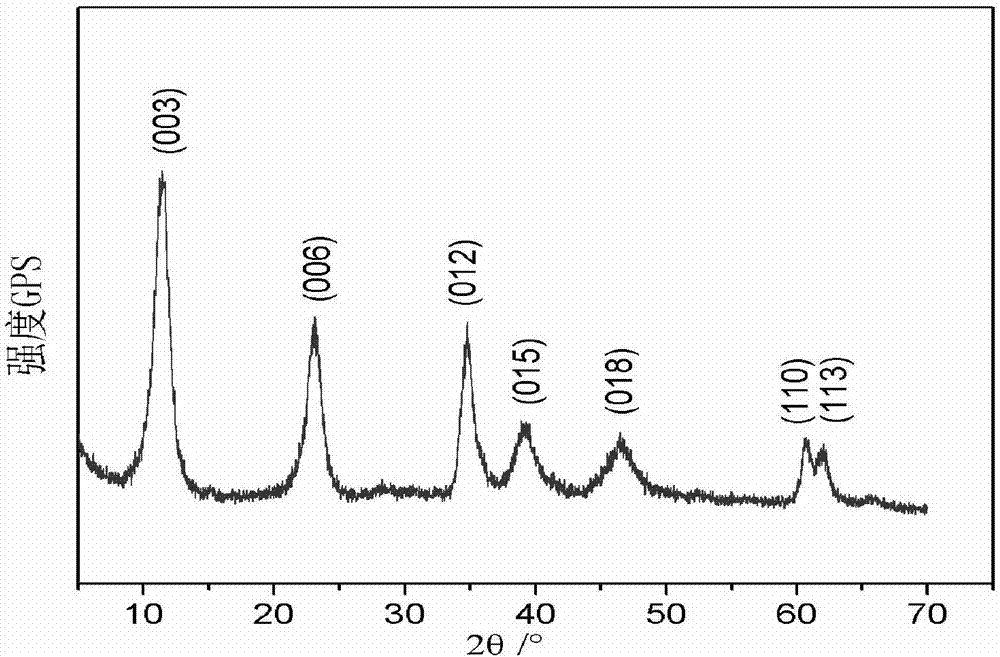
圖9為具體實施方式二中改性鎂鋁類水滑石(mgal-ldh)的xrd譜圖;
圖10為具體實施方式二中改性鎂鋁類水滑石(mgal-ldh)的ir譜圖;
圖11為具體實施方式二中純pp和5個復合材料樣品的拉伸強度柱狀圖;
圖12為具體實施方式二中純pp和5個復合材料樣品的斷裂伸長率柱狀圖;
圖13為具體實施方式二中復合材料的tga曲線;
圖14為具體實施方式二中復合材料的dtg曲線。
具體實施方式
下面結合附圖對本發明的技術方案作進一步的說明,但并不局限于此,凡是對本發明技術方案進行修改或者等同替換,而不脫離本發明技術方案的精神和范圍,均應涵蓋在本發明的保護范圍中。
具體實施方式一:本實施方式提供了一種改性鎂鋁類水滑石/聚磷酸銨/成炭發泡劑/磷腈復配阻燃劑,采用微波晶化法和共沉淀法以硝酸鋁、硝酸鎂和混合堿為原料合成鎂鋁類水滑石(mgal-ldh),然后采用硬脂酸鈉(c17h35coona)對鎂鋁類水滑石進行表面改性,并通過xrd和ft-ir對改性類水滑石樣品進行表征。將類水滑石、磷腈(cl6n3p3)、聚磷酸銨(app)和成炭發泡劑(cfa)以不同的比例復配,以形成協同膨脹阻燃劑,并以復配阻燃劑為20%的質量比和乙烯-醋酸乙烯共聚物混合聯合制成復合材料。通過阻燃性能測定、熱性能測定以及力學性能測定對復合材料的一系列性能進行研究。具體內容如下:
一、鎂鋁類水滑石(mgal-ldh)的合成及改性
采用微波晶化法制備改性鎂鋁類水滑石,具體步驟如下:
(1)分別配置1mol/l的硝酸鎂、硝酸鋁的水溶液1l。
(2)取70ml硝酸鎂和280ml硝酸鋁,得到混合鹽溶液;再取1.2mol/lna2co3和0.9mol/lnaoh混合堿600ml待用。
(3)在70℃水浴及不斷攪拌下,通過兩端的恒壓滴液漏斗向四口瓶中滴加混合鹽及混合堿。且每5秒測一次溶液ph,控制溶液ph為8-9,滴完后共沉淀反應1h。
(4)向溶液中加入0.225g硬脂酸鈉改性2h。
(5)將溶液倒入500ml燒杯中,并在70℃下微波晶化30min。
(6)用蒸餾水將溶液洗滌至ph=7,抽濾、干燥樣品。
(7)稱重裝袋。
二、類水滑石與eva混料與加工
本發明采用的是哈爾濱理工大學制造的轉矩測量儀混料,設定其混料溫度為140℃,轉速設定為60r/min,混料時間設定為10min。出料后應立即壓板,來防止產生過多氣泡,影響測定結果,取出后立即揉搓成一團,在平板硫化機上預熱3min;熱壓溫度設定為120℃,壓力10mpa,熱壓2min;之后再冷壓,力學性能測定板厚度為1mm,氧指數測定板厚度為3mm。
eva與阻燃劑總質量為180g,其中阻燃劑占復合材料總質量的20%。表1給出純eva和復合材料的配方,其中復配阻燃劑中app與cfa的質量比為3:1,通過改變類水滑石的量來考察復配阻燃劑的協同作用。
表1復合材料配方
三、結果分析
1、x-射線衍射(xrd)分析
圖1為改性鎂鋁類水滑石的xrd譜圖,本發明主要通過xrd來確定改性鎂鋁類水滑石的晶體結構與層板之間的晶面間距。由圖1可知,合成的樣品具有類水滑石結構的所有特征衍射峰,其衍射峰的2θ位于11.3°、22.7°、34.7°和60.4°處,分別對應于(003)、(006)、(009)和(110)晶面。xrd結果已表明改性鎂鋁類水滑石的合成獲得成功。并且改性鎂鋁類水滑石的d(003)值是
2、改性類水滑石的ft-ir分析
圖2為改性鎂鋁類水滑石的ir譜圖,從圖2中可以看出,樣品在3455cm-1和1638cm-1出現明顯的吸收峰。在796cm-1附近出現的肩峰則是碳酸根的面外變形振動特征峰,改性的鎂鋁類水滑石在2919cm-1和2842cm-1附近出現的吸收峰則為硬脂酸鈉的甲基、亞甲基的伸縮振動吸收峰。670cm-1附近的譜帶和1376cm-1附近出現的吸收帶分別是層間碳酸根離子的反對稱伸縮振動峰和面內彎曲振動峰,在1638cm-1附近的吸收帶歸屬于層間水分子的彎曲振動。3455cm-1附近的峰是層上的氫氧根和層間水分子的羥基伸縮振動峰。
綜合ir和xrd表征可知硬脂酸鈉不在類水滑石的層間,而是吸附在合成類水滑石顆粒的外表面,表征結果證明實驗中已成功對鎂鋁類水滑石進行表面改性。
3、阻燃劑的熱性能分析
圖3(a)-(d)分別給出了類水滑石(ldh)、聚磷酸銨(app)、成炭泡發劑(cfa)和磷腈的tg-dtg曲線。
由圖3(a)可知改性類水滑石樣品展示了兩個質量損失步驟。第一步質量損失發生在50~210℃的溫度區間內,是物理吸附水和層間水的損失。相應的在dtg曲線上的最大熱失重速率溫度分別是174℃,在第一階段的質量損失為15.0%。隨著溫度的進一步增加,第二步的質量損失發生在210~800℃的溫度區間內,主要是類水滑石表面的硬脂酸根離子和層板上的氫氧根離子,以及層間碳酸根陰離子的移除,其質量損失為35.4%。最后的剩余物質是鎂和鋁的氧化物,其剩余質量為49.6%。
由圖3(b)可知app樣品同樣展示了兩個質量損失步驟。第一步質量損失發生在50~344℃的溫度區間內,主要是氨氣、物理吸附水和層間水的損失,生成產物是聚磷酸,且在第一階段的質量損失為23.0%。在345~800℃范圍內快速分解,造成主鏈斷裂,是由焦磷酸斷裂為磷酸,同時形成多種聚磷酸恒沸物,在800℃時質量損失達71%。相應的在dtg曲線上的最大熱失重速率溫度分別是344℃和642℃,app在294℃才開始失重,故熱穩定性較高。
由圖3(c)可知cfa樣品也展示了兩個質量損失步驟。第一步質量損失發生在50~351℃的溫度區間內,是氨、物理吸附水和層間水的損失,相應的在dtg曲線上的最大熱失重速率溫度分別是307℃,在這一階段的質量損失為17.8%。隨著溫度的進一步增加,第二步的質量損失主要發生在352~800℃范圍內,是自身大分子鏈的斷裂,相應的在dtg曲線上的最大熱失重速率溫度分別是448℃,其質量損失為59.1%,最后的產物是炭,其成炭率為23.1%。
由圖3(d)可知磷腈樣品只有一個質量損失步驟。磷腈的初始降解溫度僅為88℃,在200℃時就已降解完全。對應的在dtg曲線上最大失重速率溫度為160℃。磷腈受熱分解釋放出nh3和h2s等氣體,磷腈化合物能更早的釋放出氣體,起到了隔絕氧氣的作用,可以有效阻止聚烯烴燃燒過程中產生的易燃產物與空氣中氧氣的接觸,進而起到阻燃作用。
4、類水滑石與eva復合材料的性能分析
(1)熱性能分析
圖4和圖5分別展示了含有阻燃劑為20wt.%添加量的復合材料的tga和dtg曲線。其中,圖5中曲線從上往下依次為1-8號復合材料。阻燃劑的加入導致eva復合材料在50~300℃的溫度范圍內有一個較小的質量損失,這個質量損失步驟歸因于阻燃劑中吸附水和結構水的蒸發,以及nh3和h2s等的損失。所有復合材料的質量損失過程主要發生在第二步熱降解階段,其溫度區間在300~500℃范圍內。主要是eva的醋酸基、乙烯基以及阻燃劑的降解。磷腈/eva復合材料的熱降解溫度是341℃,比純eva和其他復配阻燃劑的復合材料的熱降解溫度是351℃低,其值降低了10℃;ldh/eva復合材料的熱降解溫度是364℃,比純eva和其他復配阻燃劑復合材料的熱降解溫度是351℃有所增加,其值增大了13℃。
在dtg曲線上阻燃劑的添加使得乙烯基鏈的熱降解溫度提高到475℃,比純eva的434℃提高了41℃。添加其他阻燃劑的復合材料的乙烯基鏈的熱降解溫度均比eva有所提高。原因是由于在加熱過程中類水滑石的吸熱效應和其它阻燃劑分解釋放的nh3、h2o和co2能夠有效地延遲乙烯基鏈的裂斷。雖然四種阻燃劑的熱降解溫度各有不同,但是在高溫時乙烯基鏈的熱降解溫度基本相同,主要是由于復配阻燃劑間協同作用所致。從以上分析可知,阻燃劑的添加大幅度地提高了在高溫時乙烯基鏈的熱降解溫度。因此,所有復合材料均與純eva相比均展示了更高的熱穩定性。
(2)復合材料的阻燃性能分析
為了直觀展現阻燃劑對eva阻燃性能影響的總體趨勢,圖6給出純eva和復合材料樣品的氧指數圖。從圖6可知,樣品1純eva的氧指數是18%,2~8號復合材料的氧指數值分別是29.1%、25.0%、21.6%、30.3%、31.5%、30.4%和30.8%。
與純eva相比,添加阻燃劑的復合材料的阻燃性能均有較明顯的改善。仔細觀察能夠發現,改性鎂鋁類水滑石的添加量在復配阻燃劑中從1.6-3.2的6~8號復合材料的氧指數值大于只添加app/cfa復配阻燃劑的3-5號復合材料,并且在添加改性類水滑石的所有復合材料中是先隨著改性鎂鋁類水滑石的增加,其氧指數值變化沒有規律性,當6號復合材料中以復配阻燃劑中改性鎂鋁類水滑石添加量為1.6g,app為24g,cfa為8g和磷腈為2.4g時復合材料的氧指數達到最大,其值為31.5%,原因是由于磷腈中含有的大量氮磷鍵與其它三種阻燃劑復配起到了協同阻燃作用所致;當完全是由改性鎂鋁類水滑石作為阻燃劑的2號復合材料,其氧指數值在所有含改性類水滑石復合材料中最小,其值是29.1%。
以上分析可以得出結論,不同配比的改性鎂鋁類水滑石化合物其阻燃效果不同,且合適的配比中,以復配阻燃劑中改性鎂鋁類水滑石添加量為1.6g,app為24g,cfa為8g和磷腈為2.4g時復合材料的氧指數達到最大值為31.5%。
(3)復合材料的力學性能分析
為了直觀展現阻燃劑對eva力學性能影響的總體趨勢,圖7和圖8分別給出樣品1純eva和2-8號復合材料的的拉伸強度和斷裂伸長率數據的柱狀圖。
從圖7可以看出,樣品1純eva的拉伸強度值是19.58mpa。2~8號復合材料的拉伸強度值分別為4.0mpa、12.27mpa、17.12mpa、7.7mpa、14.27mpa、11.81mpa和13.02mpa,所有復合材料的強度均比純eva有所降低。然而,當復配阻燃劑中的改性鎂鋁類水滑石是1.6g、2.0g和3.2g時,其復合材料的拉伸強度值大于單獨含有36g改性鎂鋁類水滑石作為阻燃劑的復合材料,這些復合材料的拉伸強度值表明,適量的類水滑石的添加,對于復合材料的強度的提高起到了補強的作用。且當單獨添加磷腈阻燃劑時其拉伸強度為17.12mpa,明顯高于其他阻燃劑復合材料的拉伸強度值。
從圖8可以看出,純eva的斷裂伸長率值是1716%,2~8號復合材料的斷裂伸長率值分別為1302%、1535%、1693%、1387%、1656%、1594%和1753%。與純eva相比,當復配阻燃劑中的改性鎂鋁類水滑石是3.2g時的復合材料的拉伸強度值大于純eva。由此可見,適量的改性鎂鋁類水滑石的添加可以與app和cfa起到協同增韌的作用。而其他復合材料的拉伸強度值均小于eva。
四、結論
本實施方式合成了硬脂酸鈉改性的鎂鋁類水滑石,并將其與app、cfa和磷腈(cl6n3p3)復配成具有協同作用的膨脹阻燃劑,分別將純類水滑石和復配阻燃劑與eva制成復合材料,從而研究膨脹阻燃劑對eva的阻燃性能、力學性能和熱穩定性的影響。所得結論如下:
(1)通過xrd譜圖分析可證明,類水滑石的特征衍射峰說明均成功合成出了硬脂酸鈉表面改性的鎂鋁類水滑石。
(2)通過ft-ir分析可證明,所合成的物質是硬脂酸鈉表面改性的鎂鋁類水滑石。
(3)類水滑石、app、cfa和膦腈的熱分析測試表明,膦腈只有一步質量損失步驟,且熱降解溫度最低,僅有160℃;而其它三個阻燃劑均有兩步質量損失,app的熱降解溫度最高,其值達到642℃,cfa在所有阻燃劑中有最大的成炭率,其值為23.1%。四種阻燃劑之間的協同作用有利于eva熱穩定性和阻燃性能的提高。
(4)阻燃性能分析表明,硬脂酸鈉表面改性的類水滑石可以增加eva的阻燃性能,而將類水滑石與app、cfa和磷腈復配成特定比例的膨脹阻燃劑,其阻燃性能相對于類水滑石來說展示了較大的提高。復配阻燃劑中鎂鋁改性類水滑石添加量為1.6g,app為24g、cfa為8g和磷腈為2.4g時復合材料的氧指數達到最大,即其阻燃效果最好。
(5)力學性能分析表明,當復配阻燃劑中改性鎂鋁類水滑石添加量合適時,其復合材料的拉伸強度值明顯大于磷腈作為阻燃劑的復合材料;斷裂伸長率數據指出合適量的改性鎂鋁類水滑石的添加可以與app和cfa起到協同增韌的作用,其韌性強于純eva。
(6)熱分析分析表明,阻燃劑的添加使復合材料的熱失重速率溫度升高。從以上分析可知,阻燃劑的添加使得乙烯基鏈的熱降解溫度提高到475度,比純eva的434度提高了41度。類水滑石不僅對eva的醋酸熱降解溫度展示了提高,而且也最大的提高了在高溫時乙烯基鏈的熱降解溫度。因此,所有復合材料均比純eva有更高的熱穩定性。
具體實施方式二:本實施方式提供了一種改性鎂鋁類水滑石/聚磷酸銨/成炭發泡劑/磷腈復配阻燃劑,采用共沉淀法和微波晶化法以硝酸鎂、硝酸鋁和混合堿為原料合成鎂鋁類水滑石(mgal-ldh),然后采用硬脂酸鈉對鎂鋁類水滑石進行表面改性,并通過xrd、ft-ir和接觸角對類水滑石樣品進行表征。將類水滑石、聚磷酸銨(app)和成炭發泡劑(cfa)和磷腈(cl6n3p3)以不同的比例復配成協同膨脹阻燃劑,并以阻燃劑為20%的質量比與聚丙烯混合制成復合材料。通過氧指數和力學測試對復合材料的阻燃和力學性能進行研究。具體內容如下:
一、鎂鋁類水滑石的合成及改性
采用共沉淀法和微波晶化法制備改性鎂鋁類水滑石(mgal-ldh),具體步驟如下:
(1)分別配置1mol/l的硝酸鎂、硝酸鋁的水溶液1l。
(2)取70ml硝酸鋁及280ml硝酸鎂,得到混合鹽溶液;再取1.2mol/lna2co3和0.9mol/lnaoh混合堿500ml待用。
(3)在70℃水浴及不斷攪拌下分別由兩恒壓滴液漏斗向四口瓶中滴加混合鹽及混合堿,控制溶液ph為8~9,滴完后反應1小時。
(4)向溶液中加入0.225g硬脂酸鈉改性2小時。
(5)將溶液倒入500ml燒杯中,70℃下微波晶化30分鐘。
(6)將溶液洗滌至ph=7,抽濾、干燥樣品。
(7)稱重裝袋。
二、復配阻燃劑與聚丙烯混料與加工
采用轉矩測量儀混料,設定溫度為170℃,轉速60r/min,混料時間為10分鐘。出料后立即壓板,防止產生過多氣泡,影響測定結果,取出后在平板硫化機上壓片,熱壓溫度為150℃,熱壓時間為2min,壓力10mpa;之后再冷壓,氧指數測定板厚為3mm,力學性能測定板厚為1mm。
聚丙烯與阻燃劑總質量為180g,其中阻燃劑占復合材料總質量的20%。表2給出純聚丙烯和復合材料的配方,其中復配阻燃劑中app與cfa的質量比為3:1,通過改變類水滑石的量來考察復配阻燃劑的協同作用。
表2復合材料配方
三、結果分析
1、x-射線衍射(xrd)分析
同可見光一樣,x射線也具有波動性,因此可以發生衍射。對于固體晶態化合物來說,沒有任何兩種化合物的化學成分和晶體結構是相同的,成分和結構不同的晶體其粉末的衍射數據也是不同的。x射線粉晶衍射儀可以通過計數器和測角儀記錄衍射線的強度和方向,從而獲得衍射數據。
本實施方式主要通過xrd來確定鎂鋁類水滑石的晶體結構與層板之間的晶面間距。圖9為改性鎂鋁類水滑石的xrd譜圖,由圖9可知,合成的樣品具有類水滑石的(003)、(006)、(012)、(015)、(018)和(110)等特征衍射峰,xrd結果表明改性鎂鋁類水滑石獲得成功。并且改性鎂鋁類水滑石d(003)值分別是
2、改性類水滑石的ft-ir分析
圖10為改性鎂鋁類水滑石的ir譜圖,從圖10中可以看出,2個樣品均在3455cm-1和1638cm-1出現明顯的吸收峰。在1638cm-1附近的吸收帶是層間水分子的彎曲振動峰;1376cm-1附近的譜帶和665cm-1附近出現的吸收帶分別是層間碳酸根離子的反對稱伸縮振動峰和面內彎曲振動峰,而在796cm-1附近出現的肩峰則是碳酸根的面外變形振動特征峰,是對于改性的鎂鋁類水滑石在2919cm-1和2842cm-1附近出現的吸收峰則為硬脂酸鈉的甲基、亞甲基的伸縮振動吸收峰。由于xrd結果已證明硬脂酸鈉不在類水滑石的層間,表明硬脂酸鈉是吸附在類水滑石顆粒表面,證明實驗中對鎂鋁類水滑石的表面改性獲得成功。
3、類水滑石與聚丙烯復合材料的性能分析
(1)氧指數分析
表3給出了改性鎂鋁類水滑石-pp復合材料的氧指數數值。從表3中數據可知,含有20%改性鎂鋁類水滑石阻燃劑的復合材料的氧指數從純pp的19.3%增加到25.2%。而20%膦腈作為阻燃劑的復合材料的氧指數增加到28.9%。由類水滑石、膦腈、app和cfa復配阻燃劑的復合材料的氧指數已超過37%,特別是樣品-2復配阻燃劑中鎂鋁改性類水滑石添加量為1.6g,磷腈為2.4g,app為24g和cfa為8g時復合材料的氧指數已達到38.4%,在所有復合材料中展示了最好的阻燃性能。
表3氧指數的數值
(2)力學性能分析
為了直觀展現阻燃劑對pp力學性能影響的總體趨勢,圖11和圖12分別給出樣品1純pp和2~6號復合材料的的拉伸強度和斷裂伸長率數據的柱狀圖。從圖11可以看出,樣品1~6為純pp和5個復合材料的拉伸強度值,其值分別是24.87、28.43、25.41、18.56、20.12、19.88mpa,通過比較可知,添加1.6g和2g改性類水滑石的復合材料的拉伸強度比純pp有所提高。而含36g磷腈的復合材料表現出來的拉伸強度是最小的。拉伸強度數據顯示通過添加適當含量的改性鎂鋁類水滑石、app、cfa和磷腈等,阻燃劑間的協同作用可使得復合材料的強度好于純pp。從圖12可以看出,樣品1~6為純pp和5個復合材料的斷裂伸長率值,其值分別是19.28%、15.34%、16.93%、6.255%、14.10%、12.96%。從圖中數據可知,純pp的斷裂伸長率值在所有樣品中是最大。添加類水滑石的量為3.2g,磷腈為0.8g的復合材料的斷裂伸長率展示了最低值。
(3)熱分析
通過熱分析得到了tga、dtg兩條曲線。圖13給出了tga曲線,圖14給出了dtg曲線。
圖13和圖14分別展示了添加硬脂酸鈉表面改性的鎂鋁水滑石復合材料的tga和dtg曲線。類水滑石的加入導致pp復合材料在150~300℃的溫度范圍內有一個小的質量損失,這個損失歸因于類水滑石層間水和結構水的蒸發。所有復合材料的質量損失過程主要發生在后面兩個熱降解階段,其溫度區間在350~550℃范圍內。所有含類水滑石復配阻燃劑均提高了復合材料的碳鏈的熱降解溫度。原因是由于在加熱過程中類水滑石的吸熱效應和分解釋放的h2o和co2有效地延遲了的碳鏈裂斷。并且,類水滑石與cfa/app/磷腈體系的協同作用對提高在高溫時乙烯基鏈的熱降解溫度起了很大的作用。使得含類水滑石的復配阻燃劑阻燃的復合材料高溫下的熱穩定性均明顯強于純pp。
四、結論
(1)通過xrd譜圖分析可證明,類水滑石的特征衍射峰說明成功合成出了硬脂酸鈉表面改性的鎂鋁類水滑石。
(2)通過傅立葉紅外光譜分析可證明,所合成的化合物是硬脂酸鈉表面改性的鎂鋁類水滑石。
(3)阻燃性能分析表明,硬脂酸鈉表面改性的類水滑石可以增加pp的阻燃性能,而將類水滑石與app和cfa和磷腈復配成特定比例的膨脹阻燃劑,其阻燃性能相對于單獨類水滑石有明顯提升。復配阻燃劑中鎂鋁改性類水滑石添加量為1.6g、磷腈為2.4g、app為24g和cfa為8g時復合材料的氧指數達到最大。
(4)力學性能測試分析表明,添加app24g、cfa8g、ldh1.6g和磷腈2.4g的樣品的拉伸強度最大。
(5)含類水滑石復合材料在高溫下的熱穩定性明顯強于純pp。協效劑的加入能夠改變pp的熱降解及燃燒行為,催化促進體系在較低的溫度下分解碳化,達到更高的阻燃效果。
具體實施方式三:本實施方式與具體實施方式一、二不同的是,所述磷腈為六對羧基苯氧基環三磷腈、苯氧基磷腈、對羧基苯氧基磷腈、聚氨基磷腈或烴氧基磷腈。
具體實施方式四:本實施方式與具體實施方式一、二、三不同的是,所述naoh的濃度為0.6m,na2co3的濃度為0.45m。
- 還沒有人留言評論。精彩留言會獲得點贊!