一種基于稀土元素的溫敏性復合物及其制備方法和應用與流程
本發明涉及稀土材料,具體涉及一種基于稀土元素的溫敏性復合物及其制備方法和應用。
背景技術:
稀土高分子體系既包括稀土化合物均勻地摻雜于高分子聚合物或單體中所形成的摻雜型稀土高分子(doping-type rare earth polymers),也包括稀土離子以化學鍵合配位方式鍵合于高分子或單體中形成的鍵合型稀土高分子(bonding-type rare earth polymers),以高分子聚合物作為基質所制得的稀土高分子材料,一方面具備高分子化合物機械強度好、易加工等優點,另一方面具有稀土元素特殊的光致發光性能,因此引起了世界各國科學家的廣泛關注。
稀土元素獨特的4f電子構型使得它的化合物表現出優良的電、光、磁性能,與其他元素相比,稀土元素無法比擬的光譜學性質使得含有稀土元素的發光材料格外引人關注。目前,有關固體發光領域的研究總會不可避免的涉及到稀土發光材料。因此,研究稀土配合物以求實現它的功能化就顯得十分有意義。
聚N-異丙基丙烯酰胺,因其水溶液或水凝膠的溫度在臨界溶解溫度(LCST)附近時,它的溶脹性能和其他相關性能會發生改變,因而使它在藥物控釋體系、生物物質的分離、免疫分析、生物酶的固定等領域有潛在的應用。
綜上,通過RAFT方法將聚N-異丙基丙烯酰胺末端羧基化,制備出一種溫度響應型高分子聚合物,并將聚合物與具有熒光標記作用的稀土離子相結合,檢測其熒光特性、溫敏特性,以探求在生物工程領域應用的可行性正成為研究的方向。
技術實現要素:
本發明設計開發了一種基于稀土元素的溫敏性復合物。
本發明的第二發明目的是提供上述基于稀土元素的溫敏性復合物的制備方法。
本發明的第三發明目的是提供上述基于稀土元素的溫敏性復合物在熒光材料領域的應用以及溫敏材料領域的應用。
本發明提供的技術方案為:
一種基于稀土元素的溫敏性復合物,結構式如下:
其中,M為Eu3+或者Tb3+,n為65;以及
所述溫敏性復合物的數均分子量為Mn=7700g/mol,分子量分布為1.03,由GPC測定。
一種基于稀土元素的溫敏性復合物的制備方法,包括如下步驟:
步驟一、由α-噻吩甲酰三氟丙酮、三價稀土金屬鹽酸鹽以及5-氨基-1,10-鄰菲羅啉進行配位,得到稀土金屬配合物;
步驟二、通過1-十二硫醇、丙酮以及四丁基溴化銨作為原料,在堿性條件下,加入丙酮以及二硫化碳溶液繼續反應后,在加入氯仿和氫氧化鈉繼續作用后,酸化體系,得到S-S′-雙(1-十二烷基)三硫代醋酸酯;
步驟三、通過所述步驟二中得到S-S′-雙(1-十二烷基)三硫代醋酸酯、N-異丙基丙烯酰胺、1,3,5-三噁烷以及偶氮二異丁腈進行聚合反應,得到末端含有羧基的聚(N-異丙基丙烯酰胺);
步驟四、通過所述稀土金屬配合物與所述聚(N-異丙基丙烯酰胺)在乙醇溶液中攪拌反應,得到所述溫敏性復合物。
優選的是,在所述步驟一包括:將α-噻吩甲酰三氟丙酮溶解在乙醇中,用的氫氧化鈉調節溶液pH為6.5,將三價稀土金屬鹽酸鹽的乙醇溶液滴加到上述反應,攪拌,再加二次蒸餾水繼續恒溫攪拌,過濾,復合物在冷卻至室溫的過程中沉淀,濾出沉淀物,用水洗滌,并在真空下干燥;所得復合物為三價稀土金屬:α-噻吩甲酰三氟丙酮的摩爾數比例為1:3的二元產物,將5-氨基-1,10-鄰菲羅啉在乙醇中與所述二元產物絡合在一起,同時攪拌將溶液加熱回流,24小時后過濾,用乙醇和氯仿將過量的無界絡合物沖走,得到銪配合物。
優選的是,所述三價稀土金屬鹽酸鹽為EuCl3或者TbCl3。
優選的是,在所述步驟二中包括:將1-十二烷硫醇,丙酮與四丁基溴化銨放在三口燒瓶中反應,冷卻,在氮氣下進行混合;溶液中加入氫氧化鈉溶液混合,將反應物攪拌,再將丙酮以及二硫化碳溶液加入,要超過20分鐘,溶液顏色變成紅色,將氯仿溶液加入,然后滴加氫氧化鈉溶液超過30分鐘,將反應物攪拌過夜;溶液中加入水,然后由濃鹽酸的水溶液酸化,通過鼓動反應器,以幫助氮氣蒸發掉丙酮,收集固體,然后加入攪拌的2-丙醇,未溶解的固體濾出,得到S-S′-雙(1-十二烷基)三硫代醋酸酯。
優選的是,在所述步驟三中包括:將所述S-S′-雙(1-十二烷基)三硫代醋酸酯、N-異丙基丙烯酰胺、1,3,5-三噁烷偶氮二異丁腈放入聚合管中,加入無水N,N-二甲基甲酰胺作溶劑,放在磁力攪拌器上,進行多次通氮氣后再抽真空的操作,每次用時10~15分鐘,然后將反應混合物在氮氣保護下,放入油浴中反應,接著升溫反應,取出后冷卻,進行旋蒸,旋蒸冷卻后,加入無水乙醚沉淀,用抽濾瓶過濾,得到末端含有羧基的聚(N-異丙基丙烯酰胺)。
優選的是,在所述步驟四中包括:將所述末端含有羧基的聚(N-異丙基丙烯酰胺)和所述稀土金屬配合物放入乙醇溶液中攪拌,反應結束后將產物旋轉蒸發,除去乙醇溶液,然后將所得液體置于恒溫干燥箱中干燥后得到所述溫敏性復合物。
優選的是,所述步驟一中,合成所述5-氨基-1,10-鄰菲羅啉包括如下步驟:
步驟a、在1,10-鄰菲羅啉和濃硫酸中加入濃硝酸,放入70℃油浴鍋中,接著將溫度升到120℃恒溫攪拌,將反應混合物用冰水稀釋逐漸冷卻,同時將溫度保持在低于5℃,用堿中和到成中性,所形成的微黃色的沉淀物通過過濾收集并用冰冷的水充分洗滌,殘留物進一步用乙醇重結晶,在真空烘箱中干燥過夜后,得到5-硝基-1,10-鄰菲羅啉;
步驟b、將5-硝基-1,10-鄰菲羅啉和鈀碳催化劑的乙醇懸浮液加熱,然后通入氮氣,加入水合肼溶液,將反應混合物回流,趁熱過濾后,將濾液在旋轉式蒸發器上蒸干,將得到的復合物粗品用乙醇重結晶進一步純化,干燥后,在真空烘箱中于室溫下過夜,得到5-氨基-1,10-鄰菲羅啉。
所述的復合物在溫敏材料中的應用。
所述的復合物在熒光材料中的應用。
本發明與現有技術相比較所具有的有益效果:
1、通過本發明的合成方法所合成的DMP-PNIPAM/Eu(III)復合物最低臨界溶解溫度為34.1℃,相對于DMP-PNIPAM的最低臨界溶解溫度的33.9℃以及PNIPAM的最低臨界溶解溫度的32℃均有升高,XPS結果顯示,Eu(Ш)與配體中的O、N原子以配位鍵結合。復合物具有優異的熒光性能,熒光發射譜中580、591、614、621nm處發射峰分別對應5D0—>7fj(j=0,1,2,3,4)的電子躍遷,其中614nm處發射峰強度最大。同時測定,DMP-PNIPAM/Eu(Ш)的熒光壽命為11.78ms,使DMP-PNIPAM/Eu(III)復合物能夠作為溫敏材料以及熒光材料應用;
2、通過本發明的合成方法所合成的DMP-PNIPAM/Tb(III)復合物最低臨界溶解溫度為34.0℃,相對于DMP-PNIPAM的最低臨界溶解溫度的33.9℃以及PNIPAM的最低臨界溶解溫度的32℃均有升高,XPS結果顯示,Tb(Ш)與配體中的O、N原子以配位鍵結合。復合物具有優異的熒光性能,熒光發射譜中487、545、583、616nm處發射峰分別對應5D4—>7fj(j=6,5,4,3)的電子躍遷,其中545nm處發射峰強度最大。同時測定,DMP-PNIPAM/Eu(Ш)的熒光壽命為10.67ms,使DMP-PNIPAM/Tb(III)復合物能夠作為溫敏材料以及熒光材料應用
附圖說明
圖1為本發明所述的DMP的紅外光譜圖。
圖2為本發明所述的DMP的核磁共振氫譜圖。
圖3為本發明所述的DMP-PNIPAM的紅外光譜圖。
圖4為本發明所述的DMP-PNIPAM的核磁共振氫譜圖。
圖5為本發明所述的5-硝基-1,10-鄰菲羅啉的紅外光譜圖。
圖6為本發明所述的5-硝基-1,10-鄰菲羅啉的核磁共振氫譜圖。
圖7為本發明所述的5-氨基-1,10-鄰菲羅啉的紅外光譜圖。
圖8為本發明所述的5-氨基-1,10-鄰菲羅啉的核磁共振氫譜圖。
圖9為本發明所述的DMP-PNIPAM/Eu(III)復合物的紅外光譜圖。
圖10為本發明所述的DMP-PNIPAM/Eu(III)復合物的XPS元素分析圖。
圖11為本發明所述的DMP-PNIPAM/Tb(III)復合物的紅外光譜圖。
圖12為本發明所述的DMP-PNIPAM/Tb(III)復合物的XPS元素分析圖。
圖13為本發明所述的DMP-PNIPAM/Eu(III)復合物的熒光激發光譜圖。
圖14為本發明所述的DMP-PNIPAM/Eu(III)復合物的熒光發射光譜圖。
圖15為本發明所述的DMP-PNIPAM/Eu(III)復合物的熒光壽命圖。
圖16為本發明所述的DMP-PNIPAM/Tb(III)復合物的熒光激發光譜圖。
圖17為本發明所述的DMP-PNIPAM/Tb(III)復合物的熒光發射光譜圖。
圖18為本發明所述的DMP-PNIPAM/Tb(III)復合物的熒光壽命圖。
具體實施方式
下面結合附圖對本發明做進一步的詳細說明,以令本領域技術人員參照說明書文字能夠據以實施。
實施例1
1、合成三硫代末端羧基化合物DMP
將20mmol的1-十二烷硫醇,165.5mmol的丙酮與8mmol的四丁基溴化銨放在三口燒瓶中反應,冷卻至10℃,在氮氣下進行混合,溶液中加入21mmol的質量分數為50%的氫氧化鈉溶液,將反應物攪拌30分鐘,再將34.5mmol的丙酮和20mmol的二硫化碳混合溶液加入,要超過20分鐘,在該時間內溶液的顏色變成紅色,將30mmol的氯仿溶液加入,然后滴加100mmol的質量分數為50%氫氧化鈉溶液超過30分鐘,將反應物攪拌過夜;過夜后溶液中加入30ml水,然后加入5ml濃鹽酸水溶液酸化,通過鼓動反應器幫助氮氣蒸發掉丙酮,用布氏漏斗收集固體,將固體加入攪拌的50ml的2-丙醇中,未溶解的固體濾出,并鑒定為S-S′-雙(1-十二烷基)三硫代醋酸酯(DMP),再將2-丙醇的溶液濃縮至干,將得到的固體用正己烷中重結晶,得到4.625g黃色結晶固體,熔點62.3℃;DMP合成路線反應式如下:
2、RAFT法制備末端羧基化合物DMP-PNIPAM
將0.0508g的DMP、1.1298g的N-異丙基丙烯酰胺(PNIPAM)、0.0465g的1,3,5-三噁烷以及0.0017g的偶氮二異丁腈放入聚合管中,加入無水N,N-二甲基甲酰胺(DMF)作溶劑,放在磁力攪拌器上,循環3次通氮氣再抽真空,每次用時10分鐘,然后將反應混合物在氮氣保護下,放入60℃油浴中反應,接著將溫度升到80℃恒溫反應5h,取出冷卻,進行旋蒸除去溶劑,旋蒸完冷卻,加入無水乙醚開始沉淀,用抽濾瓶過濾,即得目標產物末端含有羧基的聚(N-異丙基丙烯酰胺)(DMP-PNIPAM);DMP-PNIPAM(A)合成路線反應式如下:
3、合成5-氨基-1,10-鄰菲羅啉
首先,在配有冷凝器和磁力攪拌棒的250mL三口燒瓶中,裝入0.017mmol的1,10-鄰菲羅啉和16mL的濃硫酸,然后加入8mL的濃硝酸,放入70℃油浴鍋中,接著將溫度升到120℃恒溫攪拌3小時后,將反應混合物用冰水稀釋逐漸冷卻至0℃,同時將溫度保持在低于5℃,用氫氧化鈉水溶液中和到成中性,所形成的微黃色的沉淀物通過過濾收集并用冰冷的水充分洗滌,殘留物進一步用體積分數為95%的乙醇重結晶,在50℃真空烘箱中干燥過夜后,得到1.3513g淺黃色固體5-硝基-1,10-鄰菲羅啉;
然后,在250ml三口燒瓶中加入19.98mmol的5-硝基-1,10-鄰菲羅啉,0.8g的質量分數為10%的Pd/C催化劑和100mL的乙醇,將該懸浮液加熱至70℃,然后通入氮氣30分鐘,加入61.5mmol的水合肼溶液,將反應混合物回流5小時,在60~70℃過濾后,將濾液在旋轉式蒸發器上蒸干,得到明亮黃色粉末的化合物粗品,將該化合物粗品用乙醇重結晶進一步純化,干燥后,在真空烘箱中于室溫下過夜,得到3.0g的黃色晶體5-氨基-1,10-鄰菲羅啉;5-氨基-1,10-鄰菲羅啉合成路線反應式如下:
4、合成稀土噻吩甲酰三氟丙酮配合物
將6mmol的α-噻吩甲酰三氟丙酮(TTA)溶解在30ml的乙醇中,用1mol·L-1的氫氧化鈉溶液調節PH≈6.5,將2mmol的EuCl3·6H2O的乙醇溶液滴加到上述反應中,60℃下攪拌20min,再加200ml二次蒸餾水繼續恒溫攪拌1h,過濾,復合物在冷卻至室溫的過程中沉淀,濾出沉淀物,用水洗滌,并在真空下干燥;所得復合物為摩爾比Eu:TTA=1:3的二元產物;
將0.1025g的5-氨基-1,10-鄰菲羅啉在乙醇中與0.1034g的Eu(TTA)3(H2O)2復合物絡合在一起,同時攪拌將溶液加熱回流,24小時后過濾,用乙醇和氯仿將過量的無界絡合物沖走,得到淺棕色產物;稀土噻吩甲酰三氟丙酮配合物(B)合成路線反應式如下:
5、合成DMP-PNIPAM/Eu(III)復合物
將DMP-PNIPAM(A)和稀土噻吩甲酰三氟丙酮配合物(B)放入乙醇溶液中攪拌,反應結束后透析(MWCO=4000),除去乙醇溶液及小分子物質,然后將所得液體置于恒溫干燥箱中24h后得到目標產物;DMP-PNIPAM/Eu(III)復合物合成路線反應式如下:
實施例2
1、合成三硫代末端羧基化合物DMP
將20mmol的1-十二烷硫醇,165.5mmol的丙酮與8mmol的四丁基溴化銨放在三口燒瓶中反應,冷卻至10℃,在氮氣下進行混合,溶液中加入21mmol的質量分數為50%的氫氧化鈉溶液,將反應物攪拌30分鐘,再將34.5mmol的丙酮和20mmol的二硫化碳混合溶液加入,要超過20分鐘,在該時間內溶液的顏色變成紅色,將30mmol的氯仿溶液加入,然后滴加100mmol的質量分數為50%氫氧化鈉溶液超過30分鐘,將反應物攪拌過夜;過夜后溶液中加入30ml水,然后加入5ml濃鹽酸水溶液酸化,通過鼓動反應器,以幫助氮氣蒸發掉丙酮,用布氏漏斗收集固體,將固體加入攪拌的50ml的2-丙醇中,未溶解的固體濾出,并鑒定為S-S′-雙(1-十二烷基)三硫代醋酸酯(DMP),再將2-丙醇的溶液濃縮至干,將得到的固體用正己烷中重結晶,得到4.625g黃色結晶固體,熔點62.3℃;DMP合成路線反應式如下:
2、RAFT法制備末端羧基化合物DMP-PNIPAM
將0.0508g的DMP、1.1298g的N-異丙基丙烯酰胺(PNIPAM)、0.0465g的1,3,5-三噁烷以及0.0017g的偶氮二異丁腈放入聚合管中,加入無水N,N-二甲基甲酰胺(DMF)作溶劑,放在磁力攪拌器上,循環3次通氮氣再抽真空,每次用時10分鐘,然后將反應混合物在氮氣保護下,放入60℃油浴中反應,接著將溫度升到80℃恒溫反應5h,取出冷卻,進行旋蒸除去溶劑,旋蒸完冷卻,加入無水乙醚開始沉淀,用抽濾瓶過濾,即得目標產物末端含有羧基的聚(N-異丙基丙烯酰胺)(DMP-PNIPAM);DMP-PNIPAM(A)合成路線反應式如下:
3、合成5-氨基-1,10-鄰菲羅啉
首先,在配有冷凝器和磁力攪拌棒的250mL三口燒瓶中,裝入0.017mmol的1,10-鄰菲羅啉和16mL的濃硫酸,然后加入8mL的濃硝酸,放入70℃油浴鍋中,接著將溫度升到120℃恒溫攪拌3小時后,將反應混合物用冰水稀釋逐漸冷卻至0℃,同時將溫度保持在低于5℃,用氫氧化鈉水溶液中和到成中性,所形成的微黃色的沉淀物通過過濾收集并用冰冷的水充分洗滌,殘留物進一步用體積分數為95%的乙醇重結晶,在50℃真空烘箱中干燥過夜后,得到1.3513g淺黃色固體5-硝基-1,10-鄰菲羅啉;
然后,在250ml三口燒瓶中加入19.98mmol的5-硝基-1,10-鄰菲羅啉,0.8g的質量分數為10%的Pd/C催化劑和100mL的乙醇,將該懸浮液加熱至70℃,然后通入氮氣30分鐘,加入61.5mmol的水合肼溶液,將反應混合物回流5小時,在60~70℃過濾后,將濾液在旋轉式蒸發器上蒸干,得到明亮黃色粉末的化合物粗品,將該化合物粗品用乙醇重結晶進一步純化,干燥后,在真空烘箱中于室溫下過夜,得到3.0g的黃色晶體5-氨基-1,10-鄰菲羅啉;5-氨基-1,10-鄰菲羅啉合成路線反應式如下:
4、合成稀土噻吩甲酰三氟丙酮配合物
將6mmol的α-噻吩甲酰三氟丙酮(TTA)溶解在30ml的乙醇中,用1mol·L-1的氫氧化鈉溶液調節PH≈6.5,將2mmol的TbCl3·6H2O的乙醇溶液滴加到上述反應中,60℃下攪拌20min,再加200ml二次蒸餾水繼續恒溫攪拌1h,過濾,復合物在冷卻至室溫的過程中沉淀,濾出沉淀物,用水洗滌,并在真空下干燥,所得復合物為摩爾比Tb:TTA=1:3的二元產物;
將0.1025g的5-氨基-1,10-鄰菲羅啉在乙醇中與0.1034g的Tb(TTA)3(H2O)2復合物絡合在一起,同時攪拌將溶液加熱回流,24小時后過濾,用乙醇和氯仿將過量的無界絡合物沖走,得到淺棕色產物;稀土噻吩甲酰三氟丙酮配合物(B)合成路線反應式如下:
5、合成DMP-PNIPAM/Tb(III)復合物
將DMP-PNIPAM(A)和稀土噻吩甲酰三氟丙酮配合物(B)放入乙醇溶液中攪拌,反應結束后透析(MWCO=4000),除去乙醇溶液及小分子物質,然后將所得液體置于恒溫干燥箱中24h后得到目標產物;DMP-PNIPAM/Tb(III)復合物合成路線反應式如下:
對實施例1以及實施例2中的復合物的結構進行表征確認。
表征方法及儀器
(1)傅里葉紅外光譜(FT-IR)測試
采用FT-IR-8400型傅里葉紅外光譜儀對合成的產物進行測試,將干燥后的樣品粉末采用KBr壓片法制樣,掃描范圍4000~500cm-1。
(2)核磁共振光譜(NMR)的測定
采用JEOL JNM-400型核磁共振儀,以CDCl3作為溶劑,以四甲基碘硅烷做為內標進行測試。
(3)紫外光譜測試
濁度法測試聚合物溫敏性是在UV mini 1240(Shimadzu)型紫外-可見分光光度計上進行的。先配制一定濃度的聚合物水溶液(2mg/mL),通過恒溫循環水槽(SDR-30,Hitachi)調節升溫速率(0.5℃/min),紫外-可見分光光度計上測定λ=500nm處聚合物水溶液的透過率,繪制透過率對不同溫度的曲線,從而確定其LCST值。
(4)熒光光譜測試
日本島津RF-5301PC型熒光光度計,激發光源為150W氙燈,分別取相同質量高分子配合物粉末置于固體樣品池中,激發及發射單色器狹縫根據樣品熒光強度均確定為3nm。
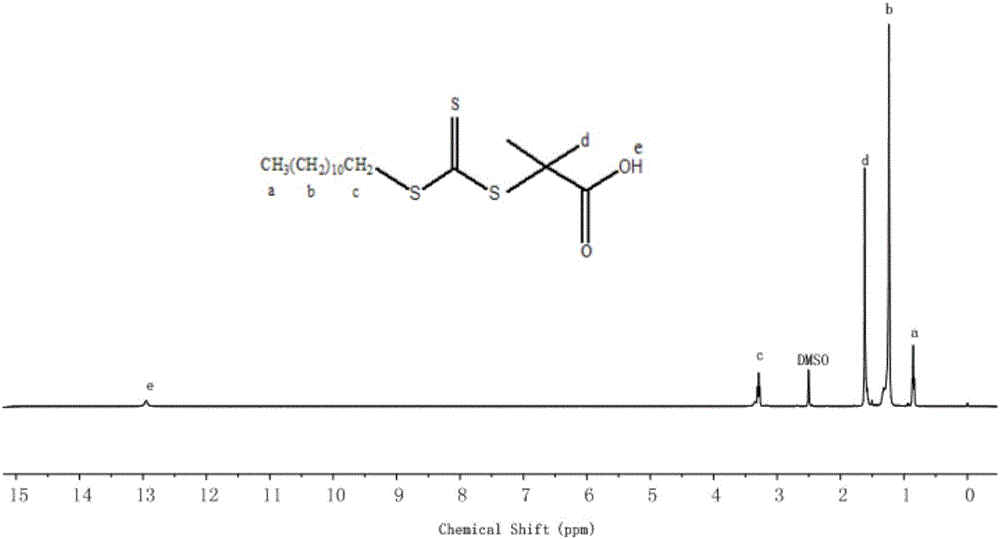
如圖1所示,3441cm-1處,為羥基的特征吸收O-H的伸縮振動特征吸收,2920cm-1、2851cm-1處分別為-CH3和-CH2-的對稱和不對稱伸縮振動峰,1719cm-1處的吸收峰為DMP中羧基C=O雙鍵伸縮振動。在吸收峰為1070cm-1處C=S鍵的吸收振動,904cm-1為甲基的C-H變形振動,812cm-1處對應于—CH2(CH2)10-CH3的面內搖擺振動;如圖2所示,圖標a處為-CH3的特征峰,圖標b處為亞甲基-CH2-的特征峰,圖標c處為與S連接的-CH2-的特征峰,圖標d處為-CH3的特征峰,圖標e為末端-COOH的特征峰;因此,綜合圖1和圖2可以判斷DMP已經生成。
如圖3所示,3304cm-1處,對應是仲氨基的N-H鍵的伸縮振動,1652cm-1為酰胺基的C=O很強的吸收譜帶,為Ⅰ譜帶,1547cm-1為N-H的彎曲振動強吸收峰,為Ⅱ譜帶;如圖4所示,分析可知,在圖標a處為十二烷基的特征峰,圖標b處為與硫連接的-CH2-的特征峰,圖標c處為是PNIPAM上重復單元次甲基上H的特征位移,圖標d處為斷裂鏈處的甲基特征峰,圖標e處為酰胺基-NH-上H的特征位移,圖標f處為聚合物PNIPAM鏈末端-CH3-上H的特征位移,圖標g處為與羧基連接碳上的甲基的特征位移;因此,綜合圖3和圖4可以判斷DMP-PNIPAM已經生成。
如圖5所示,在波數1434cm-1和1530cm-1處出現的吸收峰分別為硝基的對稱和不對稱伸縮振動特征峰,說明有-NO2的存在,在波數1600cm-1處出現的吸收峰為-C-N-的伸縮振動峰,表明鄰菲羅啉已發生硝化反應;如圖6所示,圖標a處為4C上質子H的特征峰,圖標b處為8C上質子H的特征峰,圖標c處為9C上質子H的特征峰,圖標d處為2C上質子H的特征峰,圖標e處為3C上質子H的特征峰,圖標f處為7C上質子H的特征峰,圖標g處為6C上質子H的特征峰;因此,綜合圖5和圖6可以判斷5-硝基-1,10-鄰菲羅啉已經生成。
如圖7所示,3401cm-1、3300cm-1處為氨基N-H鍵的反對稱和對稱伸縮振動特征雙峰,1640cm-1處的吸收峰對應的是C=N鍵的伸縮振動,說明有氨基存在;如圖8所示,圖標a處為4C上質子H的特征峰,圖標b處為8C上質子H的特征峰,圖標c處為9C上質子H的特征峰,圖標d處為2C上質子H的特征峰,圖標e處為3C上質子H的特征峰,圖標f處為7C上質子H的特征峰,圖標g處為6C上質子H的特征峰,圖標h處為-NH2的特征峰;因此,綜合圖7和圖8可以判斷5-氨基-1,10-鄰菲啰啉已經生成。
如圖9所示,3285cm-1處,對應是仲氨基的N-H鍵的伸縮振動,1640cm-1為酰胺基的C=O很強的吸收譜帶,為Ⅰ譜帶、1533cm-1為N-H的彎曲振動強吸收峰,為Ⅱ譜帶。相比DMP-PNIPAM紅外光譜圖對應的相應吸收峰,均發生了紅移現象,說明Eu3+可能與PNIPAM側鏈的氮原子或羰基氧發生配位,吸收電子的誘導效應分別使NH,-C=O鍵間電子云密度下降,鍵力常數變小,致使振動頻率降低,導致紅位移現象;如圖10、表1所示,曲線a為DMP-PNIPAM的XPS全圖,曲線b為DMP-PNIPAM/Eu(III)的XPS全圖,結合能在530.3eV、399.1eV、284.2eV處的峰值表明了復合物中的O、N、C元素的存在,曲線b與a相比,多了Eu的峰值,從曲線c上,141.5eV處的峰值代表了Eu元素的存在,DMP-PNIPAM/Eu(Ш)與配體DMP-PNIPAM相比,它的O1s、N1s結合能都有所增加,而DMP-PNIPAM/Eu(Ш)配合物中Eu(Ш)的4d的結合能比EuCl3中Eu(Ш)的4d的結合能低。由于內層電子的屏蔽作用受核外電子的電荷分布影響,因此當外層電荷密度降低時,內層電子的屏蔽作用就會減弱,內層電子的結合能隨之增加,與此相反結合能便會減小。因此可以推斷DMP-PNIPAM/Eu(Ш)中配體DMP-PNIPAM羧基上的氧原子在形成配位鍵后電子轉移到稀土Eu(III)的外層空軌道上,使得Eu(Ш)外層電子密度增加,屏蔽作用增強,Eu(Ш)電子結合能下降,在誘導效應的作用下氮原子將電子傳遞給氧原子,造成氮原子外層的電子云密度減小,屏蔽作用減弱,而內層電子結合能升高。XPS測試結果反映了配體與Eu(Ш)的配位成鍵情況,表明已經成功合成了Eu(III)與DMP-PNIPAM的復合物。
表1EuCl3、DMP-PNIPAM、DMP-PNIPAM/Eu(Ш)的O1s、N1s和Tb4d的結合能
如圖11所示,在3286cm-1處為N-H鍵的伸縮振動峰,在1641cm-1處為NH-C=O的C=O很強的吸收譜帶,在1532cm-1處為N-H的彎曲振動強吸收峰。相比DMP-PNIPAM紅外光譜圖對應的相應吸收峰,均發生了紅移現象。這種紅移現象可能是由于稀土噻吩甲酰三氟丙酮配合物中-NH2基與DMP-PNIPAM中的-COOH基發生酰化反應引起的,也有可能稀土噻吩甲酰三氟丙酮配合物并不穩定,分解出的Tb3+與PNIPAM側鏈的氮原子或羰基氧發生配位,吸收電子的誘導效應分別使N-H,C=O鍵間電子云密度下降,鍵力常數變小,致使振動頻率降低,導致紅移;如圖12、表2所示,DMP-PNIPAM/Tb(Ш)與配體DMP-PNIPAM相比,它的O1s結合能有增加了1.74eV,同時配合物中的N1s結合能增加了2.57eV,而DMP-PNIPAM/Tb配合物中Tb(Ш)的4d的結合能比TbCl3中Tb(Ш)的4d的結合能低4.29eV,表明DMP-PNIPAM與Tb(Ш)反應成功。由于內層電子的屏蔽作用受核外電子的電荷分布影響,因此當外層電荷密度降低時,內層電子的屏蔽作用就會減弱,內層電子的結合能隨之增加,與此相反結合能便會減小。因此可以推斷DMP-PNIPAM/Tb(Ш)中配體DMP-PNIPAM羧基上的氧原子在形成配位鍵后電子轉移到稀土Tb(III)的外層空軌道上,使得Tb(Ш)外層電子密度增加,屏蔽作用增強,Tb(Ш)電子結合能下降,在誘導效應的作用下氮原子將電子傳遞給氧原子,造成氮原子外層的電子云密度減小,屏蔽作用減弱,而內層電子結合能升高。
表2TbCl3、DMP-PNIPAM、DMP-PNIPAM/Tb(Ш)的O1s、N1s和Tb4d的結合能
對實施例1中的DMP-PNIPAM/Eu(III)復合物以及實施例2中的DMP-PNIPAM/Tb(III)復合物進行GPC分析;
凝膠滲透色譜通過具有分子篩性質的固定相,用來分離相對分子質量較小的物質,并且還可以分析分子體積不同、具有相同化學性質的高分子同系物。
GPC的色譜條件為:流速為1.0ml/min,柱溫15.0℃,進樣量20μL,視差折光檢測器。流動相為四氫呋喃,用Agilent HP1100專用GPC軟件采集和分析數據。用凝膠滲透色譜法測定分子量時,由于非體積效應等原因無法進行直接測量,需要找到適合的色譜條件,制定正確的校正曲線才能有效克服非體積效應。
GPC測定DMP-PNIPAM/Eu(III)復合物中聚合物的Mn=7700g/mol,多分散性指數PDI(Mw重均/Mn數均)為1.03,DMP的分子量為364,N-異丙基丙烯酰胺分子量為113.18,經計算可知,聚合度n=65,轉化率46.3%(產量/反應物的量)。
GPC測試DMP-PNIPAM/Tb(III)復合物中聚合物的Mn=7700g/mol,多分散性指數PDI(Mw重均/Mn數均)為1.03,DMP的分子量為364,N-異丙基丙烯酰胺分子量為113.18,經計算可知,聚合度n=65,轉化率為46.3%(產量/反應物的量)。
試驗例
1、熒光性能檢測
如圖13、圖14、圖15所示,首先,以620nm的發射波長測銪復合物的激發光譜,在以275nm光激發,測得銪復合物的發射光譜,圖13在270—370nm出現了3個與激發光譜峰重疊的半頻峰,圖14在620nm附近出現了兩個倍頻峰,這兩項干擾峰均與次配合物的熒光光譜性無關;在275nm紫外光激發下,位于580和590nm處出現兩組強度不同的發射峰;由圖14可知:(1)三元配合物體系發射譜中僅出現Eu3+離子5D0—>7fj(j=0,1,2,3,4)躍遷對應的特征發射峰,分別位于580nm,591nm,614nm,621nm,其中614nm處5D0—>7f2電偶極躍遷發射峰發射強度最強,為配體微擾的中心離子的發光;(2)DMP-PNIPAM/Eu(III)三元配合物只發射Eu3+的特征熒光,單色性好,色純度高;(3)PNIPAM在325nm處的發射峰在三元配合物中幾乎完全消失,可能是高分子的激發三重態與Eu3+的激發態能級之間差別較大,不能進行有效的能量傳遞,使高分子吸收的能量以非輻射方式損耗;如圖15所示,經過熒光測定,測得銪復合物的熒光壽命達到11.78ms,其量子產率為Φ=0.176,DMP-PNIPAM聚合物本身不具有熒光性,但是加入很少部分Eu(Ш)形成銪的復合物后,發出紅色熒光,壽命相對來說很長,因此,本發明所述的DMP-PNIPAM/Eu(III)復合物可以作為熒光材料的應用。
如圖16、圖17、圖18所示,DMP-PNIPAM/Tb(III)復合物的熒光激發光譜特征峰強度在350nm處最大,這一結果說明在高分子聚合物中加入稀土鋱(III)和小分子配體TTA,復合物同樣具有熒光特性,TTA的加入與Tb(III)粒子發生了協同配位作用,更大程度地滿足Tb(III)的配位數,這種方式也大大提高了Tb(III)離子的發射效率,使復合物熒光性能增強,從圖17可知,復合物體系發射譜中僅出現Tb3+離子5D4—>7Fj(j=6,5,4,3)躍遷對應的特征發射峰,分別位于487nm,545nm,583nm,616nm,其中545nm處5D4—>7F5電偶極躍遷發射峰發射強度最強,為配體微擾的中心離子的發光,DMP-PNIPAM/Tb(III)復合物只發射Tb3+的特征熒光,單色性好,色純度高,PNIPAM在325nm處的發射峰在復合物中幾乎完全消失,可能是高分子的激發三重態與Tb3+的激發態能級之間差別較大,不能進行有效的能量傳遞,使高分子吸收的能量以非輻射方式損耗;圖16、圖17充分的證明了配位成功,即聚合物具有熒光性;由圖18可知,DMP-PNIPAM/Tb(III)復合物的熒光壽命達到10.67ms,因為DMP-PNIPAM聚合物本身不具有熒光性,但是加入很少部分Tb(Ш)的配合物后,發出綠色熒光,并且能達到10.67ms,熒光壽命較長,由此可以推斷,合成出熒光性能較好的DMP-PNIPAM/Tb(Ш)復合物;因此,本發明所述的DMP-PNIPAM/Tb(III)復合物可以作為熒光材料的應用。
2、溫敏性檢測
如表3所示,聚N-異丙基丙烯酰胺的低臨界溶解溫度(LCST)為32℃左右,而DMP-PNIPAM的地臨界溶解溫度(LCST)為33.9℃,相對于聚N-異丙基丙烯酰胺的臨界溶解溫度有所提高,這是因為鄰苯二甲酸二甲酯與聚N-異丙基丙烯酰胺聚合后,分子上有極性基團羧基的緣故。DMP-PNIPAM/Eu(III)復合物的地臨界溶解溫度為34.1℃,少量Eu(Ш)的加入可使DMP-PNIPAM的LCST略微升高。這是由于DMP-PNIPAM/Eu(III)聚合物溶液中Eu(Ш)離子配位數較高還可以與水分了配位,稀土離子與水分子羥基配位鍵的鍵能高于水分子本身氫鍵的鍵能,因此當溫度升高至LCST附近時,需要消耗更多的能量才能破壞稀土離子與水分子間的配位鍵,使配合物發生構象轉變,從而使高分子配合物的相轉變略微滯后;因此,本發明所述的DMP-PNIPAM/Eu(III)復合物可以作為溫敏材料的應用。
表3DMP-PNIPAM和DMP-PNIPAM/Eu(III)復合物的臨界溶解溫度(LCST)
如表4所示,將制備好的DMP-PNIPAM和DMP-PNIPAM/Tb(III)復合物用紫外-可見光光譜分析,由于在聚合過程中PNIPAM末端引入了親水性的羧基,使得DMP-PNIPAM的LCST較PNIPAM(32℃)溫度略微升高。少量Tb(Ш)的加入可使DMP-PNIPAM的LCST溫度略微升高。這是由于DMP-PNIPAM/Tb(III)聚合物溶液中Tb(Ш)離子配位數較高還可以與水分子配位,稀土離子與水分子羥基配位鍵的鍵能高于水分子本身氫鍵的鍵能,因此當溫度升高至LCST溫度附近時,需要消耗更多的能量才能破壞稀土離子與水分子間的配位鍵,使配合物發生構象轉變,從而使高分子配合物的相轉變略微滯后;因此,本發明所述的DMP-PNIPAM/Tb(III)復合物可以作為溫敏材料的應用。
表4DMP-PNIPAM和DMP-PNIPAM/Tb(III)復合物的臨界溶解溫度(LCST)
盡管本發明的實施方案已公開如上,但其并不僅僅限于說明書和實施方式中所列運用,它完全可以被適用于各種適合本發明的領域,對于熟悉本領域的人員而言,可容易地實現另外的修改,因此在不背離權利要求及等同范圍所限定的一般概念下,本發明并不限于特定的細節和這里示出與描述的圖例。
- 還沒有人留言評論。精彩留言會獲得點贊!