通過二硫代碳酸酯或其硒類似物與雙酚的酯交換制備聚碳酸酯的方法與流程
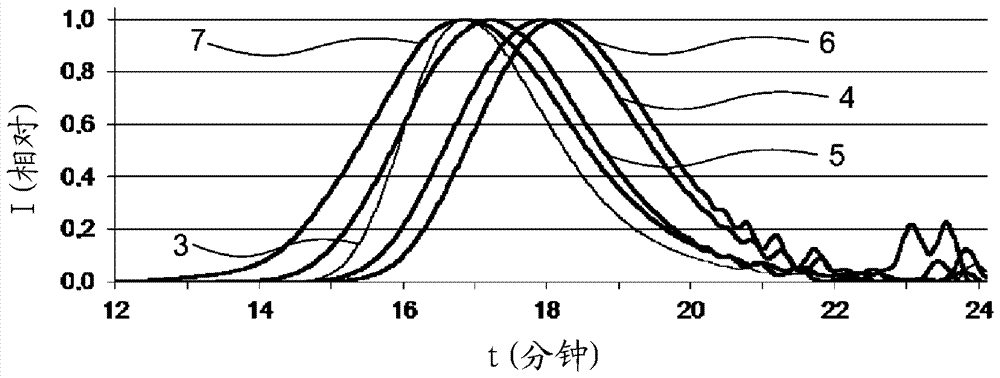
本發明涉及制備芳族聚碳酸酯的方法,其包括使雙酚與二硫代碳酸酯或其硒類似物在催化劑存在下反應的步驟。本發明還涉及二硫代碳酸酯或其硒類似物作為用于制備聚碳酸酯的酯交換試劑的用途。基于雙酚a的聚碳酸酯在技術和工業上非常有意義。聚碳酸酯通常通過根據相界面法的雙酚a光氣化或通過碳酸二芳基酯,特別是碳酸二苯酯與雙酚a在熔體中的催化酯交換制備。在相界面法中,在多級反應中,光氣在至少一種溶劑中與芳族二醇的堿性水溶液反應。作為在有機溶劑中的溶液獲得聚碳酸酯。聚合物溶液的后處理包括溶劑的再循環,以產生廢水料流。例如在us5,399,659和us5,340,905中描述了根據熔體酯交換法制備聚碳酸酯。由芳族二醇,例如雙酚a和碳酸二芳基酯,例如碳酸二苯酯通過使用各種催化劑和使用不同的溫度和壓力制備芳族聚碳酸酯,以連續除去生成的副產物。熔體酯交換法中所用的碳酸二芳基酯可在商業上通過碳酸二烷基酯與酚的酯交換制備。例如,ep0781760a1描述了通過使碳酸二烷基酯與芳族羥基化合物在催化劑存在下反應制備芳族碳酸酯和連續除去在反應中形成的碳酸二芳基酯、醇類副產物、碳酸二烷基酯和芳族羥基化合物的連續方法,其中將碳酸二烷基酯和芳族羥基化合物再循環到該反應中。wo-a2004/016577a1描述了在反應器裝置的多個單獨和串聯的反應區中在催化劑存在下由碳酸二烷基酯和芳族羥基化合物制備碳酸二芳基酯的方法,其中利用在最后一個反應區的蒸氣料流的冷凝中獲得的冷凝熱來加熱引入第一反應區的液體料流。但是,這種方法的缺點是復雜的反應器裝置。此外,這種方法的能量集成需要改進并且僅限于反應的工藝段。沒有描述隨后的后處理步驟。碳酸二烷基酯與酚直接反應的缺點是酚式oh基團被該碳酸酯烷基化的趨勢。在碳酸二甲酯與酚的反應中情況尤其如此,以致形成相應的甲基醚。這一副反應尤其描述在catal.commun.33(2013)20–23中。因此,碳酸二烷基酯沒有直接用于雙酚a的縮聚以產生芳族聚碳酸酯,而是首先與單酚反應以產生碳酸二芳基酯。這例如在asahi法中實現,其詳細描述在greenchem.5(2003)497-507中。通過使碳酸的酯或硫代酸酯與烷撐二醇反應來制備脂族聚碳酸酯是wo97/03104a1中已知的。本發明解決的問題因此是提供制備芳族聚碳酸酯的改進的方法,其中與相界面法相比,需要較少的反應和后處理步驟并且其中在縮聚中形成的副產物比在現有技術描述的反應的情況下更容易分離。根據本發明,通過制備聚碳酸酯的方法解決該問題,其包括使雙酚與酯交換試劑在催化劑存在下反應的步驟,其中所述酯交換試劑包含通式(i)的化合物:其中x和x'各自獨立地為s或se,優選s,且r和r'各自獨立地為烷基或芳基或r和r'一起是亞烷基鏈。本發明的方法提供制備芳族聚碳酸酯而無副反應的優點,而在通過脂族碳酸酯的酯交換制備芳族聚碳酸酯中酚的烷基化是顯著的副反應。可以從反應混合物或最終產物中通過蒸餾除去在該反應中形成的硫醇或硒醇。本發明的方法同樣具有短鏈二硫代碳酸s,s'-二烷基酯在室溫下是液體的優點,而碳酸二芳基酯是固體。通過液態的物態,簡化該混合物在反應開始時的加工,因為酚可溶解在二硫代碳酸s,s'-二烷基酯中。基于二硫代碳酸酯的制備聚碳酸酯的方法不僅在聚碳酸酯的制備方面有利,而且在該二硫代碳酸酯的制備方面也有利。例如,可以與碳酸二烷基酯類似地通過相應烷硫醇化合物的光氣化來制備二硫代碳酸二烷基酯。同樣已知的是借助硒由co和相應的硫醇通過使用催化劑來合成環狀二硫代碳酸酯(tetrahedronlett.34(1974)2899-2900)和非催化合成線性二硫代碳酸酯(synlett10(2005)1535-1538)。式(i)的二硫代碳酸酯的基于光氣的制備相對于碳酸二芳基酯的基于光氣的制備而言是有利的,因為該硫代化合物尤其具有較低熔點、較低摩爾質量和較小摩爾體積。式(i)的二硫代碳酸酯的無光氣制備相對于碳酸二芳基酯的已知無光氣制備而言是有利的,因為省略從碳酸二烷基酯產生碳酸二芳基酯的酯交換步驟。不同于碳酸二烷基酯,二硫代碳酸二烷基酯可以直接用于制備聚碳酸酯而不經酯交換成碳酸二芳基酯。在本發明中,術語“聚碳酸酯”包括低聚和聚合的聚碳酸酯化合物。通過本發明的方法獲得的聚合聚碳酸酯優選具有18000至80000克/摩爾,更優選19000至50000克/摩爾的數均分子量mn。可以通過在thf中對照聚苯乙烯標樣借助凝膠滲透色譜法測量而測定數均分子量。在通式i中,x和x'各自獨立地為s或se,優選s。合適的通式(i)的化合物的實例是其中r和r'各自獨立地為直鏈或支化、任選取代的c1-c34-烷基或任選取代的c5-c34-芳基,優選c1-c6-烷基或c6-c10-芳基,更優選c1-烷基、c2-烷基或c6-芳基的那些。r和r'可以相同或不同。r和r'優選相同。r和r'基團也可以一起是任選取代的c2-c12-亞烷基鏈。式(i)中的c1-烷基是甲基,c2-烷基是乙基,且c6-芳基是苯基。式(i)中的c1-c6-烷基另外是例如正丙基、異丙基、正丁基、仲丁基、叔丁基、正戊基、1-甲基丁基、2-甲基丁基、3-甲基丁基、新戊基、1-乙基丙基、環己基、環戊基、正己基、1,1-二甲基丙基、1,2-二甲基丙基、1-甲基戊基、2-甲基戊基、3-甲基戊基、4-甲基戊基、1,1-二甲基丁基、1,2-二甲基丁基、1,3-二甲基丁基、2,2-二甲基丁基、2,3-二甲基丁基、3,3-二甲基丁基、1-乙基丁基、2-乙基丁基、1,1,2-三甲基丙基、1,2,2-三甲基丙基、1-乙基-1-甲基丙基或1-乙基-2-甲基丙基;c1-c34-烷基另外是例如正庚基和正辛基、頻哪基(pinakyl)、金剛烷基、異構的薄荷基、正壬基、正癸基、正十二烷基、正十三烷基、正十四烷基、正十六烷基或正十八烷基。這同樣適用于例如芳烷基或烷基芳基中的相應烷基。相應羥基烷基或芳烷基/烷基芳基中的亞烷基是例如與上述烷基對應的亞烷基。芳基是具有5至34個環原子的碳環芳族、任選被雜原子取代的基團。這同樣適用于芳基烷基(也稱作芳烷基)的芳族部分,以及更復雜的基團,例如芳基羰基的芳基成分。芳基烷基或芳烷基各自獨立地代表如上文定義的直鏈、環狀、支化或非支化的烷基,其可以被如上定義的芳基單取代、多取代或全取代。特別優選的通式(i)的化合物是二硫代碳酸二甲酯、二硫代碳酸二乙酯和二硫代碳酸二苯酯。式(i)的化合物可以以與碳酸二烷基或二芳基酯對應的方式制備。用于制備碳酸二烷基或二芳基酯的已知方法是例如在壓力下無溶劑的氣相光氣化或液相光氣化(ep2586787a1)、相界面光氣化、氧化羰基化(us2003055199a)、直接(wo2008044575a1)或通過環狀碳酸酯的酯交換(例如wo2007069529、wo2008065874)羧基化。適用于制備碳酸二烷基和二芳基酯的催化劑是已知的并可用于制備式(i)的化合物。根據本發明,根據式(i)的酯交換試劑與雙酚,優選式(ii)的雙酚反應以制備聚碳酸酯:其中r5和r6各自獨立地代表h、c1-c18-烷基-、c1-c18-烷氧基、鹵素如cl或br或各自任選取代的芳基或芳烷基,優選h或c1-c12-烷基,更優選h或c1-c8-烷基,最優選h或甲基,且z代表單鍵、-so2-、-co-、-o-、-s-、c1-c6-亞烷基、c2-c5-烷叉基或c6-c12-亞芳基,它們可以任選與含雜原子的其它芳環稠合。可用于本發明的方法的此類化合物的實例是二羥基二芳基烷,如氫醌、間苯二酚、二羥基聯苯、雙(羥苯基)鏈烷、雙(羥苯基)環烷、雙(羥苯基)硫醚、雙(羥苯基)醚、雙(羥苯基)酮、雙(羥苯基)砜、雙(羥苯基)亞砜、α,α'-雙(羥苯基)二異丙基苯和它們的烷基化、在環上烷基化和在環上鹵化的化合物。雙酚的實例是4,4'-二羥基聯苯、2,2-雙(4-羥苯基)-1-苯基丙烷、1,1-雙(4-羥苯基)苯基乙烷、2,2-雙(4-羥苯基)甲烷(雙酚f)、2,2-雙(4-羥苯基)丙烷(雙酚a(bpa))、2,4-雙(4-羥苯基)-2-甲基丁烷、1,3-雙[2-(4-羥苯基)-2-丙基]苯(雙酚m)、2,2-雙(3-甲基-4-羥苯基)丙烷、雙(3,5-二甲基-4-羥苯基)甲烷、2,2-雙(3,5-二甲基-4-羥苯基)丙烷、雙(3,5-二甲基-4-羥苯基)砜、2,4-雙(3,5-二甲基-4-羥苯基)-2-甲基丁烷、1,3-雙[2-(3,5-二甲基-4-羥苯基)-2-丙基]苯、1,1-雙(4-羥苯基)環己烷和1,1-雙(4-羥苯基)-3,3,5-三甲基環己烷(雙酚tmc)。特別優選的雙酚是4,4'-二羥基聯苯、1,1-雙(4-羥苯基)苯基乙烷、2,2-雙(4-羥苯基)甲烷(雙酚f)、2,2-雙(4-羥苯基)丙烷(雙酚a(bpa))、2,2-雙(3,5-二甲基-4-羥苯基)丙烷、1,1-雙(4-羥苯基)環己烷和1,1-雙(4-羥苯基)-3,3,5-三甲基環己烷(雙酚tmc)。合適的雙酚也描述在us2999835a、us3148172a、us2991273a、us3271367a、us4982014a和us2999846a、德國公開說明書de1570703a、de2063050a、de2036052a、de2211956a和de3832396a、法國專利fr1561518a、h.schnell的專著chemistryandphysicsofpolycarbonates,intersciencepublishers,newyork1964,第28頁及其后;第102頁及其后和d.g.legrand,j.t.bendler,handbookofpolycarbonatescienceandtechnology,marceldekkernewyork2000,第72頁及其后中。優選地,式(i)的化合物和雙酚以至少1:1,更優選1:1至2:1的摩爾比用于本發明的方法。合適的催化劑的實例是無機或有機的堿性化合物,例如選自氫氧化物、碳酸鹽、鹵化物、酚鹽、二酚鹽、氟化物、乙酸鹽、磷酸鹽、磷酸氫鹽和硼酸鹽(boranate)的鋰鹽、鈉鹽、鉀鹽、銫鹽、鈣鹽、鋇鹽和鎂鹽,和另外氮堿和磷堿,例如四甲基氫氧化銨、四甲基乙酸銨、四甲基氟化銨、四苯基硼酸四甲基銨、四苯基氟化鏻、四苯基硼酸四苯基鏻、二甲基二苯基氫氧化銨、四乙基氫氧化銨、1,8-二氮雜雙環[5.4.0]十一-7-烯(dbu)、1,5-二氮雜雙環[4.3.0]壬-5-烯(dbn),和胍體系,例如1,5,7-三氮雜雙環[4.4.0]癸-5-烯、7-苯基-1,5,7-三氮雜雙環[4.4.0]癸-5-烯、7-甲基-1,5,7-三氮雜雙環[4.4.0]癸-5-烯、7,7'-亞己基二-1,5,7-三氮雜雙環[4.4.0]癸-5-烯、7,7'-亞癸基二-1,5,7-三氮雜雙環[4.4.0]癸-5-烯、7,7'-亞十二烷基二-1,5,7-三氮雜雙環[4.4.0]癸-5-烯,和磷腈,例如叔辛基亞氨基三(二甲基氨基)正磷(磷腈堿p1-t-oct)、叔丁基亞氨基三(二甲基氨基)正磷(磷腈堿p1-t-butyl)和2-叔丁基亞氨基-2-二乙基氨基-1,3-二甲基全氫-1,3,2-二氮雜-2-正磷(bemp),和含磷的籠形分子,如三烷基斷鍵磷雜氮三環(prophosphatrane),例如三甲基-、三乙基-、三異丁基-、三芐基-或三-4-甲氧基芐基-斷鍵磷雜氮三環和質子海綿,如1,8-雙(二甲基氨基)萘或1,8-雙(六甲基三氨基磷腈基)萘。這些催化劑優選以基于1摩爾雙酚計1至10-8摩爾的量使用。該催化劑也可以以兩種或更多種催化劑的組合使用。在反應完成后,該催化劑可以留在產物中、分離、中和或掩蓋。該(一種或多種)催化劑優選留在產物中。可通過根據本發明的方法獲得的芳族聚碳酸酯可以通過使用少量支化劑以受控方式支化。合適的支化劑是:間苯三酚、4,6-二甲基-2,4,6-三(4-羥苯基)庚烷、1,3,5-三(4-羥苯基)苯、1,1,1-三(4-羥苯基)乙烷、三(4-羥苯基)苯基甲烷、2,2-雙[4,4-雙(4-羥苯基)環己基]丙烷、2,4-雙(4-羥苯基異丙基)苯酚、2,6-雙(2-羥基-5'-甲基芐基)-4-甲基苯酚、2-(4-羥苯基)-2-(2,4-二羥苯基)丙烷、正對苯二甲酸六(4-(4-羥苯基異丙基)苯基)酯(orthoterephthalates?ureester)、四(4-羥苯基)甲烷、四(4-(4-羥苯基異丙基)苯氧基)甲烷、靛紅雙甲酚(isatinbiskresol)、季戊四醇、2,4-二羥基苯甲酸、苯均三酸、氰尿酸、1,4-雙(4',4''-二羥基三苯基)甲基)苯和α,α',α''-三(4-羥苯基)-1,3,4-三異丙烯基苯。特別優選的是1,1,1-三(4-羥苯基)乙烷(thpe)和靛紅雙甲酚(tbk)。這些支化劑可以在該方法的每個任意階段中加入。可以例如在低分子量聚碳酸酯制備中的第一階段中進行該添加。也可以在低分子量聚碳酸酯的熔體酯交換以產生高分子量聚碳酸酯中的第二階段中進行該添加。根據本發明獲得的聚碳酸酯可含有鏈終止劑。相應的鏈終止劑尤其從ep335214a和de3007934a中獲知。鏈終止劑的實例是單酚,如苯酚,烷基苯酚,如甲酚、對叔丁基苯酚、對-正辛基苯酚、對-異辛基苯酚、對-正壬基苯酚和對-異壬基苯酚,鹵代苯酚,如對氯苯酚、2,4-二氯苯酚、對溴苯酚和2,4,6-三溴苯酚,以及對枯基苯酚。特別優選的是苯酚和對枯基苯酚。下面描述本發明的實施方案和其它方面。它們可以任意互相組合,除非從上下文中清楚看出相反的意思。在本發明的方法的一個優選實施方案中,連續除去在反應過程中形成的硫醇和/或硒醇。尤其在使用二硫代碳酸s,s'-二甲基酯和/或二硫代碳酸s,s'-二乙基酯(其中該反應產生甲硫醇和/或乙硫醇)的情況下,可以通過蒸餾除去氣態硫醇實現該除去。在使用二硫代碳酸s,s'-二苯基酯(其中該反應產生苯硫酚)的情況下,同樣可以通過蒸餾除去苯硫酚實現該除去。在本發明的方法的另一實施方案中,該反應在第一溫度和第一壓力下進行第一時期,然后在第二溫度和第二壓力下進行第二時期,其中另外地,第二溫度高于第一溫度且第二壓力低于第一壓力。不受制于理論,但推測,在該反應的第一步驟中,主要形成低分子量聚碳酸酯,在第二步驟中,這些低分子量聚碳酸酯縮合形成高分子量聚碳酸酯。在該反應的第二階段中的溫度提高和壓力降低可以各自獨立地一次性、在幾個步驟中或以分級方式進行。尤其包括多級法,其中該反應在進一步的溫度和進一步的壓力下進行至少一個進一步的時期,等等。優選地,這一進一步的方法步驟中的溫度高于前兩個溫度,且壓力低于前兩個壓力。優選地,第一溫度為150℃至210℃且第二溫度為210℃至400℃。更優選地,第一溫度為175℃至205℃。與此獨立地,250℃至350℃的第二溫度是優選的。同樣優選地,第一壓力為200毫巴至900毫巴且第二壓力為1毫巴至200毫巴。更優選地,第一壓力為250毫巴至600毫巴。與此獨立地,≤10毫巴的第二壓力是優選的。例如,該聚碳酸酯合成可以如下通過二硫代碳酸酯與雙酚在熔體中的酯交換進行:在第一階段中,雙酚和催化劑與二硫代碳酸酯在170至210℃的溫度下在900至200毫巴,優選600至400毫巴的減壓下在0.01至3小時內熔融,以形成低分子量低聚物。在該反應的第二階段中,在210至400℃,優選250至350℃的溫度和最高10毫巴的減壓下在0.01至5小時內進行高分子量聚碳酸酯的制備。本發明的方法可以例如在攪拌釜、薄層蒸發器、降膜蒸發器、攪拌釜級聯、擠出機、捏合機、簡單盤式反應器或高粘度盤式反應器中不連續或連續地進行。該方法優選連續進行。在第二溫度和第二壓力下進行第二時期的反應也可以在蒸發擠出機、盤式反應器或蒸發壓延機中進行。在本發明的方法的另一實施方案中,所用雙酚是雙酚a、雙酚f和/或雙酚tmc。在本發明的方法的另一實施方案中,通式(i)中的r和r'是甲基、乙基或苯基。特別優選的是二硫代碳酸二甲酯。在本發明的方法的另一實施方案中,通式(i)的化合物是二硫代碳酸二烷基酯,優選二硫代碳酸二甲酯或二硫代碳酸二乙酯。在本發明的方法的另一實施方案中,催化劑是鏻鹽或雙環胺。優選的鏻鹽是通式(iii)的那些其中r1、r2、r3和r4可以各自獨立地為相同或不同的c1-至c18-亞烷基、c6至c10-芳基或c5至c6-環烷基,且x-可以是陰離子,其中相應的酸-堿對h++x-→hx具有<11的pkb值。特別優選的鏻鹽是四苯基氟化鏻、四苯基硼酸四苯基鏻和四苯基苯酚鏻,尤其是四苯基苯酚鏻。特別優選的催化劑是四苯基苯酚鏻和dbu。在本發明的方法的另一實施方案中,該催化劑在反應過程中分多份加入。例如,該催化劑可以分三等份加入。在本發明的方法的另一實施方案中,其進一步包括將碳酸二芳基酯或碳酸二烷基酯添加到所得反應產物中的步驟。在一個可選的實施方案中,在式(i)的化合物與雙酚的反應開始時或過程中加入碳酸二芳基酯或碳酸二烷基酯。由此,可以進一步提高預先獲得的聚碳酸酯的分子量。此外,降低該聚碳酸酯中的硫代酸酯基團和oh基團的含量。所用碳酸二芳基酯優選是碳酸二苯酯(dpc)。碳酸二芳基酯或碳酸二烷基酯的量可以例如是原始使用的雙酚量的5摩爾%至15摩爾%。本發明還涉及通式(i)的化合物作為用于制備聚碳酸酯的酯交換試劑的用途:其中x和x'是s或se,優選s,且r和r'各自獨立地為烷基或芳基或r和r'一起是亞烷基鏈。關于r和r'的定義,參考與本發明的方法相關的上述注解。優選使用二硫代碳酸s,s'-二烷基酯,尤其是二硫代碳酸s,s'-二甲酯或二硫代碳酸s,s'-二乙酯,或二硫代碳酸s,s'-二芳基酯,尤其是二硫代碳酸s,s'-二苯基酯作為酯交換試劑。實施例通過下列實施例和對比例詳細例示本發明,但不限于此。所用酯交換試劑:二硫代碳酸二甲酯二硫代碳酸s,s'-二甲酯碳酸二甲酯碳酸o,o'-二甲酯所用雙酚:雙酚a2,2-雙(4-羥苯基)丙烷用作參比物質的聚碳酸酯:makrolon來自bayermaterialscienceag的基于雙酚a的芳族均聚碳酸酯,其具有mn=13900克/摩爾的數均分子量和2.4的分散度。雙酚a與二硫代碳酸二甲酯的聚合反應產生線性聚碳酸酯,其含有式(xa)中所示的游離oh基團和/或式(xb)中所示的不對稱硫代酸酯和/或式(xc)中所示的烷基化酚基團作為可能的端基。在添加碳酸二苯酯的情況下(參見實施例3),獲得另外或僅含式(xd)中所示的單官能酚基團作為可能的端基的聚碳酸酯線性聚合分子通常含有式(xa)至(xd)的兩個端基,其中各分子可含有兩個相同或兩個不同的端基。將各樣品溶解在氘代氯仿中并在bruker波譜儀(av400,400mhz)上測量。借助1hnmr波譜法測定雙酚a的官能化程度。為此,將不同共振的強度互相積分。用于積分的1hnmr波譜中的相關共振(基于tms=0ppm)如下:i1:1.59:雙酚a重復單元的ch3基團i2:2.32:不對稱硫代酸酯(xb)的ch3基團i3:3.69:烷基化酚(xc)的ch3基團i4:6.57-6.60和6.96-6.99:雙酚a上的和具有直接鍵合的游離oh基團的末端雙酚a單元(xa)的苯基環的芳族ch基團i5:7.07-7.09和7.16-7.18:基于雙酚a的重復單元上的和沒有直接鍵合的游離oh基團的末端雙酚a單元(xa)的苯基環的芳族ch基團。雙酚a的官能化程度的數值基于所用雙酚a的轉化的酚基團的含量并以摩爾%給出。考慮到相對強度,該值如下計算:官能化程度=i5/(i4+i5)。借助凝膠滲透色譜法(gpc)測定所得聚合物的數均分子量mn和重均分子量mw。遵循din55672-1:"凝膠滲透色譜法,第1部分–四氫呋喃作為洗脫液"(psspolymerservice的securitygpcsystem,流速1.0ml/min;柱:2×psssdvlinearm,8×300mm,5μm;rid檢測器)進行。已知摩爾質量的聚苯乙烯樣品用于校準。在具有套裝的回流冷凝器的schlenk管中進行雙酚與酯交換試劑的反應。schlenk管用調節到指定溫度的電加熱罩從外部加熱。借助存在于反應混合物中的磁攪拌棒攪拌該反應混合物。實施例1:雙酚a與二硫代碳酸二甲酯在一步法中反應在具有套裝的回流冷凝器的100毫升schlenk管中向雙酚a(5.71克,25.0毫摩爾)中加入二硫代碳酸二甲酯(3.05克,25.0毫摩爾)和四苯基苯酚鏻(216毫克,基于所用碳酸酯計2.0摩爾%)。隨后,該反應混合物在750毫巴的壓力下用電加熱罩加熱至200℃6小時。獲得6.62克粘性材料。在芳族氫區域(5.5-8.5ppm)中的1hnmr分析顯示雙酚a的75%官能化。產物的gpc分析得出mn=570克/摩爾的數均分子量和3.53的多分散度。實施例2:雙酚a與二硫代碳酸二甲酯在兩步法中反應在具有套裝的回流冷凝器的100毫升schlenk管中向雙酚a(5.71克,25.0毫摩爾)中加入二硫代碳酸二甲酯(4.88克,40.0毫摩爾)和四苯基苯酚鏻(85毫克,基于所用碳酸酯計2.0摩爾%)。隨后,該反應混合物在兩個階段中加熱。首先,將該混合物在500毫巴的壓力下在回流下加熱至250℃1小時。隨后,將回流冷凝器換成蒸餾橋。將該反應混合物繼續加熱至250℃,在此過程中首先使氬氣流經過反應混合物1小時,然后將壓力降至10毫巴30分鐘。隨后,關閉該容器并將反應混合物在<1毫巴的壓力下加熱至300℃2小時。在此過程中,淺棕色沉淀物升華在容器的上壁上。沒有測定收率。在芳族氫區域(5.5-8.5ppm)中的1hnmr分析顯示雙酚a的完全官能化。產物的gpc分析得出mn=1700克/摩爾的數均分子量和1.94的多分散度。實施例3:雙酚a與二硫代碳酸二甲酯在兩步法中反應并加入碳酸二苯酯以降低產物中的硫代酸酯基團和oh基團的含量聚合在具有套裝的回流冷凝器的100毫升schlenk管中向雙酚a(5.71克,25.0毫摩爾)中加入二硫代碳酸二甲酯(4.27克,35.0毫摩爾)和四苯基苯酚鏻(76毫克,基于所用碳酸酯計0.5摩爾%)。隨后,將反應混合物分階段加熱。首先,將該混合物在400毫巴的壓力下在回流下加熱至220℃1小時。隨后,將回流冷凝器換成蒸餾橋。將反應混合物在10毫巴的壓力下加熱1小時,在每種情況下在20分鐘后將溫度從220℃逐步提高到260℃,然后到300℃。加入碳酸二芳基酯或碳酸二烷基酯現在加入碳酸二苯酯(dpc,428毫克,2毫摩爾)。隨后,關閉該容器并將反應混合物在<1毫巴的壓力下加熱至300℃1小時。在芳族氫區域(5.5-8.5ppm)中的1hnmr分析顯示在第一反應步驟后雙酚a的85%官能化。產物的1hnmr分析在所有方面都符合makrolon。產物的gpc分析得出mn=11300克/摩爾的數均分子量和4.340的多分散度。獲得5.62克聚碳酸酯(87.8%收率)。實施例4(對比例):雙酚a與碳酸二甲酯反應在具有套裝的回流冷凝器的100毫升schlenk管中向雙酚a(5.71克,25.0毫摩爾)中加入碳酸二甲酯(3.15克,35.0毫摩爾)和四苯基苯酚鏻(76毫克,基于所用碳酸酯計0.5摩爾%)。隨后,該反應混合物在400毫巴的壓力下加熱。觀察到強回流;在燒瓶中同樣形成白色固體。該反應混合物的溫度在1小時內升至73℃的最大溫度。通過在芳族氫區域(5.5-8.5ppm)中的產物混合物1hnmr分析表明該反應混合物主要(>60%)含有未轉化的雙酚a以及未轉化的碳酸二甲酯。表1:本發明的實施例1-3與對比例4的結果的比較實施例方法雙酚a的官能化程度[%]所得產物的分子量[克/摩爾]多分散度1一步法755703.532兩步法10017001.943兩步法100113004.344(對比)一步法40--實施例1至3與對比例4的比較表明,在使用二硫屬化物碳酸酯(dichalkogenidcarbonat)作為酯交換試劑的情況下獲得低聚聚碳酸酯(實施例1)或聚合聚碳酸酯(實施例2和3),而在使用碳酸二甲酯作為酯交換試劑的情況下(對比例4),雙酚a與碳酸二甲酯僅以小程度反應并且沒有獲得聚碳酸酯。實施例5:雙酚a與二硫代碳酸二甲酯在兩步法中反應在具有套裝的回流冷凝器的100毫升schlenk管中向雙酚a(5.71克,25.0毫摩爾)中加入二硫代碳酸二甲酯(4.88克,40.0毫摩爾)和四苯基苯酚鏻(85毫克,基于所用碳酸酯計0.5摩爾%)。隨后,將反應混合物分階段加熱。首先,將該混合物在400毫巴的壓力下在回流下加熱至220℃1小時。隨后,將回流冷凝器換成蒸餾橋。將反應混合物在10毫巴的壓力下加熱1小時,在每種情況下在20分鐘后將溫度從220℃逐步提高到260℃,然后到300℃。隨后,關閉該容器并將反應混合物在<1毫巴的壓力下加熱至300℃1小時。獲得5.85克聚碳酸酯(91.4%收率)。產物的gpc分析得出mn=10000克/摩爾的數均分子量和2.92的多分散度。在芳族氫區域(5.5-8.5ppm)中的1hnmr分析顯示在第一反應步驟后雙酚a的90%官能化。產物的1hnmr分析在所有重要方面都符合makrolon。與makrolon比較的所得聚碳酸酯的凝膠滲透色譜圖(gpc)顯示在圖1中。曲線1是makrolon,曲線2是本發明的聚合物樣品。實施例6:雙酚a與二硫代碳酸二甲酯在兩步法中反應在具有套裝的回流冷凝器的100毫升schlenk管中向雙酚a(5.71克,25.0毫摩爾)中加入二硫代碳酸二甲酯(4.88克,40.0毫摩爾)和1,8-二氮雜雙環[5.4.0]十一-7-烯(dbu,60毫克,基于所用碳酸酯計1.0摩爾%)。隨后,將反應混合物分階段加熱。首先,將該混合物在500毫巴的壓力下在回流下加熱至250℃1小時。隨后,將回流冷凝器換成蒸餾橋。將反應混合物在10毫巴的壓力下加熱1小時,在此過程中首先將反應混合物加熱至250℃半小時,然后加熱至300℃另外半小時。隨后,關閉該容器并將反應混合物在<1毫巴的壓力下加熱至300℃1小時。獲得6.31克聚碳酸酯(98.6%收率)。產物的gpc分析得出mn=3700克/摩爾的數均分子量和2.37的多分散度。在芳族氫區域(5.5-8.5ppm)中的1hnmr分析顯示在第一反應步驟后雙酚a的90%官能化。產物的1hnmr分析在所有重要方面都符合makrolon。比較實施例催化劑所用催化劑的量[摩爾%]*雙酚a的官能化程度[%]所得產物的分子量[克/摩爾]多分散度3四苯基苯酚鏻2.0100113004.345四苯基苯酚鏻0.5>95100002.926dbu1.0~9537002.37*基于碳酸酯計的摩爾%。實施例3、5和6的比較表明,對于二硫屬化物碳酸酯與雙酚a的反應,可以使用四芳基鏻芳基化物(實施例3和5)和雙環胺化合物(實施例6)作為催化劑。實施例7:雙酚a與二硫代碳酸二甲酯在兩步法中反應在具有套裝的回流冷凝器的100毫升schlenk管中向雙酚a(5.71克,25.0毫摩爾)加入二硫代碳酸二甲酯(4.88克,40.0毫摩爾)和四苯基苯酚鏻(85毫克,基于所用碳酸酯計0.5摩爾%)。隨后,將反應混合物分階段加熱。首先,將該混合物在750毫巴的壓力下在回流下加熱至200℃1小時。隨后,將回流冷凝器換成蒸餾橋。將反應混合物在10毫巴的壓力下加熱1小時,在此過程中將溫度在200℃下保持30分鐘,然后逐步提高到250℃,然后300℃各15分鐘。隨后,關閉該容器并將反應混合物在<1毫巴的壓力下加熱至300℃1小時。獲得5.56克聚碳酸酯(86.9%收率)。產物的gpc分析得出mn=6500克/摩爾的數均分子量和2.48的多分散度。在芳族氫區域(5.5-8.5ppm)中的1hnmr分析顯示在第一反應步驟后雙酚a的80%官能化。產物的1hnmr分析在所有重要方面都符合makrolon。比較實施例第一階段第二階段第三階段所得產物的分子量[克/摩爾]多分散度5400毫巴,220℃10毫巴,220→300℃<1毫巴,300℃100002.927750毫巴,200℃10毫巴,200→300℃<1毫巴,300℃65002.48實施例5和7的比較表明,對于二硫屬化物碳酸酯與雙酚a的反應可以使用不同溫度和壓力等級。圖2顯示makrolon(曲線3)和在不添加dpc的情況下由雙酚a與二硫代碳酸二甲酯的聚合(實施例5)在各種反應時間后(曲線4:2小時,曲線5:3小時)獲得的反應產物和由在2小時反應時間后添加dpc的情況下的反應(實施例3)在各種反應時間下(曲線6:2小時,在臨添加dpc前;曲線7:3小時,在完全聚合后)獲得的反應產物的凝膠滲透色譜圖的比較。明顯顯示出由于dpc添加造成的分子量提高。當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1