用于嵌段共聚物的取向控制的氟?醇添加劑的制作方法
背景
本發明涉及用于定向自組裝用途中所用的嵌段共聚物的取向控制的氟-醇添加劑,更具體涉及用于含聚碳酸酯嵌段的高chi(χ)嵌段共聚物的頂部取向控制的材料。
嵌段共聚物(bcps)在溶液、本體和薄膜中用于許多用途。bcps的薄膜用途由于一些bcps形成形態部尺寸為5納米至50納米的周期性自組裝結構的能力而對納米光刻和圖案化特別有吸引力。bcps的薄膜自組裝性質可以與現有光刻技術一起使用以提供用于半導體用途的獲得長程有序的獨特途徑。被稱作嵌段共聚物的定向自組裝(dsa)的這一途徑有望擴展傳統光刻法的圖案化能力。
用于定向自組裝(dsa)用途的bcps包含兩種或更多種聚合物嵌段,它們可以相分離成以球體、圓柱體、螺旋二十四面體(gyroids)和片層的有序納米陣列為特征的結構域。bcp的相分離能力取決于該嵌段共聚物的floryhuggins相互作用參數chi(χ)和聚合程度。縮寫為ps-b-pmma的聚(苯乙烯)-b-聚(甲基丙烯酸甲酯)是最廣泛用于dsa的嵌段共聚物。對于ps-b-pmma,聚(苯乙烯)嵌段(ps)和聚(甲基丙烯酸甲酯)嵌段(pmma)在升高的溫度下在聚合物-空氣界面處具有類似的表面能。在這種情況下,將布置在取向控制層上的bcp薄層退火引發相分離以產生與該取向控制層垂直取向的bcp結構域。通常,對于ps-b-pmma,該取向控制層是用苯乙烯和甲基丙烯酸甲酯形成的可交聯或刷型無規共聚物。可以通過調節苯乙烯和甲基丙烯酸甲酯的組成控制該取向控制層(墊層)的中性潤濕性質以使bcp層狀結構域能夠垂直取向。
由于ps和pmma之間的較低相互作用和相互作用參數chi(χ),ps-b-pmma的最小半節距限于大約10納米。通常當該嵌段共聚物的chi和n的乘積小于10時,這一嵌段共聚物傾向于形成無序聚集體而非有序的相分離結構域。為了能使形態部(feature)進一步小型化(即使用具有較低聚合度的嵌段共聚物),在兩個嵌段之間具有較高相互作用參數(較高chi)的嵌段共聚物是合意的。已經研究了數種具有兩個嵌段之間的較高相互作用參數的嵌段共聚物以獲得較小形態部尺寸。特別有意義的是包含衍生自來自第一聚合物嵌段上的反應性端基的環狀羰基單體的開環的嵌段的嵌段共聚物。通過環狀單體(例如丙交酯、內酯、環氧乙烷)的開環聚合(rop)形成的嵌段共聚物已用于生成用于圖案化用途的亞10納米形態部尺寸。
隨著嵌段共聚物的兩個嵌段之間的相互作用參數提高,由于這兩個嵌段的聚合物-空氣表面能之間的失配提高,僅靠中性墊層性質可能不足以形成垂直取向的嵌段共聚物結構域。這造成嵌段共聚物結構域的平行取向,在聚合物-空氣界面僅存在較低表面能嵌段,以使該薄膜不適合光刻用途。頂涂層基取向控制策略已被用于控制嵌段在聚合物-空氣界面處的表面能。但是,這些策略在高chi嵌段共聚物集成到標準光刻法中引入額外的工藝復雜性。
需要開發用于使具有亞10納米半節距的高chi嵌段共聚物的嵌段共聚物結構域垂直取向的無頂涂層法。
概述
因此,公開了一種組合物,其包含:
i)溶劑;
ii)嵌段共聚物,其包含:
a)包含式(b-1)的重復單元的第一嵌段:
其中rw是選自h、甲基、乙基和三氟甲基(*-cf3)的一價基團且rd是包含連接到碳1上的芳環的一價基團,和
b)連接到第一嵌段上的脂族聚碳酸酯第二嵌段(聚碳酸酯嵌段);和
iii)包含氟-醇重復單元(hfa重復單元)的表面活性聚合物(sap),所述hfa重復單元包含至少一個六氟異丙基醇基團(hfa基團),所述hfa基團具有結構
其中所述嵌段共聚物和所述sap溶解在所述溶劑中。
還公開了一種方法,其包括:
提供包含頂層(墊層)的第一分層結構;
將權利要求1的組合物布置在所述墊層上并除去所有溶劑,由此形成包含布置在所述墊層上的膜層的第二分層結構,其中所述膜層包含非共價締合的所述嵌段共聚物和所述sap,且所述膜層具有與氣氛接觸的頂表面;和
允許或誘發所述膜層的嵌段共聚物自組裝,由此形成包含具有特征性節距lo的相分離結構域圖案的第三分層結構,所述結構域圖案包含交替結構域,該交替結構域含有各自的所述嵌段共聚物的化學上不同的嵌段;
其中
各結構域與所述墊層和氣氛接觸,且
包含聚碳酸酯嵌段的結構域具有比包含所述嵌段共聚物的第一嵌段的結構域高的sap濃度。
公開了另一方法,其包括:
提供包含頂層(墊層)的第一分層結構;
形成布置在所述墊層上的形貌抗蝕圖案,所述抗蝕圖案包含具有凹區的形態部(features),所述凹區具有作為所述墊層的表面的一部分的底表面;
將權利要求1的組合物基本布置在所述抗蝕圖案的凹區中并除去所有溶劑,由此形成包含膜層的第二分層結構,所述膜層包含所述嵌段共聚物和所述sap,其中所述膜層與所述底表面接觸,所述底表面對所述嵌段共聚物中性潤濕,且所述膜層具有與氣氛接觸的頂表面;和
允許或誘發所述嵌段共聚物自組裝,由此形成在所述凹區中包含所述嵌段共聚物的相分離結構域圖案(結構域圖案)的第三分層結構,其中各結構域與所述底表面和氣氛接觸,且各結構域垂直于所述墊層的主平面取向。
還公開了式(h-2)的表面活性聚合物(sap):
e'-p'-e″(h-2),
其中
e'是第一端基,
e″是第二端基,
e'和/或e″包含1-25個氟,且
p'是包含式(h-1)的氟-醇重復單元(hfa重復單元)的聚合物鏈:
其中
a'是1或2,
q″是具有a'+1的化合價的連接基,且q″包含至少一個碳,且
r″是選自*-h、甲基(*-ch3)、乙基(*-et)和三氟甲基(*-cf3)的一價基團。
本領域技術人員從下列詳述、附圖和所附權利要求書會認識到和理解本發明的上述和其它特征和優點。
附圖的若干視圖的簡述
圖1a至1c顯示在示意1至3中在二嵌段共聚物的自組裝后二嵌段共聚物的嵌段a和b的排列。
圖2.1a至2.1f是顯示使用包含所公開的含聚碳酸酯嵌段的高chi嵌段共聚物和所公開的表面活性聚合物(sap)的自組裝層(sa層)形成垂直取向的層狀結構域圖案的方法的橫截面層圖。該墊層對該嵌段共聚物中性潤濕。
圖2.2a至2.2e是顯示在形貌預圖案存在下形成垂直取向的層狀結構域圖案的方法的橫截面層圖。在不存在sap的情況下,空氣界面對該嵌段共聚物不是中性潤濕的。在自組裝過程中空氣界面變得對該嵌段共聚物中性潤濕。抗蝕形態部對包含高chi嵌段共聚物和sap的sa層可以是非中性潤濕。
圖3-84分別是實施例92-177的自組裝嵌段共聚物膜的原子力顯微術(afm)高度圖像。各afm圖像用相應的實施例號標記。
圖85a和85b是用實施例178形成的自組裝嵌段共聚物膜在不同放大率下的掃描電子顯微(sem)圖像。
圖86a–86c是用實施例180和實施例181形成的自組裝嵌段共聚物膜在不同放大率下的sems。
圖87-89分別是實施例188-190的自組裝嵌段共聚物膜的afm高度圖像。
圖90-91分別是實施例194-195的自組裝嵌段共聚物膜的afm高度圖像。
圖92-96分別是實施例200-204的自組裝嵌段共聚物膜的afm高度圖像。
圖97-103分別是實施例205-211的自組裝嵌段共聚物膜的afm高度圖像。
圖104-105分別是實施例214-215的自組裝嵌段共聚物膜的afm高度圖像。
圖106a和106b分別是實施例102的自組裝嵌段共聚物膜的自頂向下和橫截面sem圖像。
圖106c是在實施例216中通過對實施例102的自組裝嵌段共聚物膜進行gisaxs(掠入射小角度x射線散射)分析獲得的在傅里葉空間中繪制的yoneda帶上方的背景校正積分散射強度的曲線圖。
圖107-112是分別顯示實施例227-232的自組裝嵌段共聚物膜的sims(二次離子質譜學)分析的曲線圖。
詳述
公開了用于利用嵌段共聚物的自組裝的光刻法的相選擇性和表面活性聚合物(saps)。saps在下文中也被稱作“氟-醇添加劑”或“聚合物添加劑”。saps提供通過包含聚碳酸酯嵌段的高chi嵌段共聚物(bcps)的自組裝形成的相結構域的取向控制。“高chi”是指bcps具有大的flory-huggins相互作用參數chi(χ)。chi參數越高,不同bcp嵌段與彼此的溶混性越差,并且在自組裝后分離含有bcp的不同嵌段的相結構域的相界越清晰。saps優先可溶于通過自組裝形成的聚碳酸酯結構域(即包含嵌段共聚物的聚碳酸酯嵌段的結構域)。與不含sap的其它方面相同的聚碳酸酯結構域相比,saps還降低聚碳酸酯結構域的表面能,由此使聚碳酸酯結構域能夠潤濕氣氛界面,以產生具有垂直取向的片層和圓柱體。
在本文中,“氣氛”是與sa層的頂表面接觸的在任何合適壓力下的的氣體,可包括空氣和/或一種或多種其它氣體。
“sa材料”是能夠自組裝成在組成上不同的相分離結構域的材料。自組裝(sa)是指sa材料在合適的條件下發生相分離以產生不溶混固相結構域的圖案的過程。自組裝可在形成sa層時自發發生或其可以誘發(例如通過將包含sa材料的sa層在升高的溫度下退火合適的時間)。sa材料優選是嵌段共聚物(bcp)。
用于自組裝的嵌段共聚物包含至少兩種彼此不溶混的嵌段。非限制性的嵌段共聚物包括二嵌段和三嵌段共聚物。嵌段共聚物的自組裝通過嵌段的相分離發生以形成分離的固相結構域的圖案。根據嵌段的體積分數,結構域可具有片層、球體、圓柱體和/或螺旋二十四面體的形式。作為一個實例,二嵌段共聚物的自組裝可產生包含基本含有二嵌段共聚物的第一嵌段a的第一層狀結構域和基本含有二嵌段共聚物的第二嵌段b的第二層狀結構域的結構域圖案。在這種情況下,第一和第二層狀結構域通過使嵌段共聚物的嵌段a和嵌段b連接的共價鍵連接。
在本文中,“sa層”是包含sa材料和sap的層。將sa層布置在基底的頂表面上。sa層可包含其它材料。
在本文中,與sa層的底部接觸的基底的頂表面的任何材料統稱為“墊層材料”或“取向控制材料”。包含墊層材料的層是“墊層”或“取向控制層”。墊層表面影響sa層的sa材料的自組裝。
如果所提到的結構域可優先于自組裝sa材料的另一結構域潤濕表面,該表面被說成對自組裝sa材料的該提到的結構域具有“優先親和性”或對其“優先”。否則,該表面被說成對所提到的結構域“無優先”。
基底是在其上布置sa層的分層結構。基底可包含一個或多個堆疊布置的材料層,更尤其是用于制造半導體器件的材料。作為非限制性實例,基底可包括底層(例如硅片、金屬箔)、硬掩模層、介電層、金屬氧化物層、氧化硅層、氮化硅、氮化鈦、氧化鉿、抗反射層(arc)和/或用于自組裝的取向控制層(墊層)。在基底的頂表面(其通常是取向控制層的頂表面)上布置sa層。當在該取向控制層上形成抗蝕圖案時,基底包括抗蝕圖案。在這種情況下,可以在抗蝕圖案的溝槽中布置sa層。
基底的頂表面可以是用于自組裝的“制圖外延(graphoepitaxial)預圖案”或“化學預圖案”。各類型的預圖案可以由形貌形態部(topographicalfeatures)構成,如在抗蝕圖案中。制圖外延預圖案通過預圖案的形貌和表面性質影響自組裝。“化學預圖案”主要通過預圖案的不同區域的表面性質影響自組裝。在這兩類預圖案之間不存在清晰的尺寸界限,因為形貌對自組裝的影響程度還取決于sa層相對于下方浮凸表面的厚度。但是,一般而言,當使用制圖外延預圖案時,sa層的厚度小于或等于預圖案的形貌形態部的高度。對于化學預圖案,sa層厚度大于預圖案的下方形貌形態部的任何高度。
術語“界面”是指兩個基本不溶混相之間的接觸邊界。各相可以獨立地為固體、液體或氣體。
層狀或圓柱形結構域可平行或垂直于下方取向控制層(墊層)的平面或sa層的主平面取向。當層狀結構域的主平面或板(plate)平行于下方表面(或sa層)的主平面取向時,層狀結構域具有平行取向。當層狀結構域的主平面或板垂直于下方表面(或sa層)的主平面取向時,層狀結構域具有垂直取向。當圓柱軸平行于下方表面(或sa層)的主平面取向時,圓柱形結構域具有平行取向。當圓柱軸垂直于下方表面(或sa層)的主平面取向時,圓柱形結構域具有垂直取向。
層狀結構域的垂直取向對通過給定層狀結構域的選擇性蝕刻形成高分辨率線條圖案是合意的。平行取向對形成高分辨率線條圖案不合意。
術語“布置”是指層與另一層的表面接觸。除非另行指明,“布置”或“施加”是指形成與另一層的表面接觸的層,而不限制所用方法,只要獲得布置或施加的層的合意特征,如均勻性和厚度。
術語“澆鑄”是指通過在表面上布置溶解在溶劑中的材料溶液并除去溶劑而形成材料層。
無規共聚物通過名稱中的“-co-”或“-r-”指示。嵌段共聚物通過名稱中的“-b-”或“-嵌(block)-”指示。交替嵌段共聚物通過名稱中的“-alt-”指示。
在本文中,“對稱潤濕”是指墊層表面和氣氛界面被自組裝sa材料的相同結構域潤濕。“不對稱”潤濕是指墊層表面和氣氛界面被自組裝sa材料的不同結構域潤濕。
在本文中,如果自組裝sa材料的各結構域在自組裝后與一表面或氣氛界面具有接觸,表面或氣氛界面被說成對sa材料是“中性”的或相對于sa材料“中性潤濕”。否則,表面或氣氛界面被說成對sa材料“非中性”。例如,如果在自組裝后該自組裝嵌段共聚物的各結構域與墊層表面具有接觸,則墊層表面對嵌段共聚物“中性潤濕”。應該理解的是,自組裝sa材料的各結構域可包含sap,且各結構域在自組裝后可具有不同sap濃度。中性墊層表面允許取向控制但不引導自組裝結構域的橫向空間排列。非中性墊層表面可引導自組裝。對本發明而言,墊層和氣氛最好對通過包含sa材料和sap的sa層的自組裝形成的結構域中性潤濕是合意的。
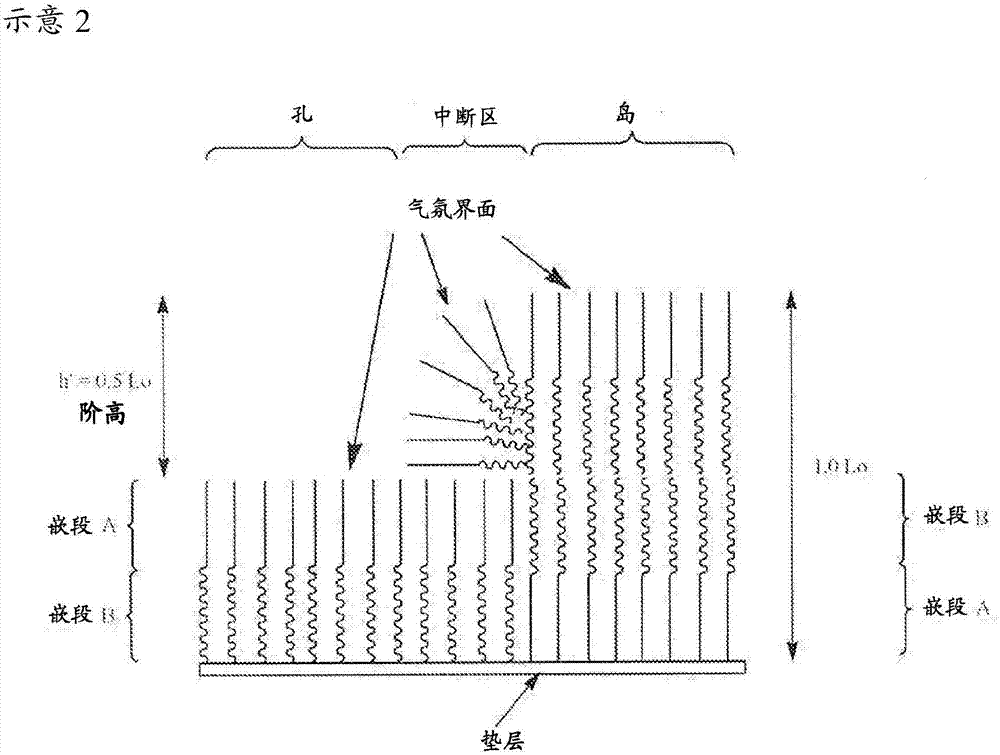
對于商業用途,墊層表面和氣氛界面必須對sa材料無優先(中性)以在沒有其它化學或形貌形態部可用于影響嵌段共聚物的自組裝時獲得垂直取向的層狀結構域。如果僅一個界面對sa材料中性,則層狀結構域平行于墊層表面取向以形成具有0.5lo(“l零”)階高的島/孔狀態。“階高”是指相對于周圍sa材料的高度差,且lo是自組裝sa材料的結構域的特征性節距(體相周期性(bulkperiodicity))。sa材料的chi參數越高,結構域圖案的潛在lo(節距)越小。
這些參數圖示在下文進一步給出的示意1-3的示意中。如果僅墊層表面是中性的,兩種嵌段共聚物(bcp)結構域都最初以0.5lo垂直片層潤濕墊層表面,但由于氣氛(例如空氣)非中性而最終形成平行狀態,以產生包含具有0.5lo階高的平行片層的島/孔。
為了證實本發明,基底包含布置在硅片上的取向控制層(墊層)。墊層表面可以是具有均勻表面性質的平坦表面(即墊層表面沒有形貌或化學圖案化)。下述實施例大多使用平坦墊層。另一些實施例使用在墊層上具有形貌抗蝕圖案以用于制圖外延法(graphoepitaxy)的基底。sa層布置在墊層上并具有與氣氛接觸的頂表面。合意的是包含sa材料和sap的sa層的自組裝形成包含與墊層和氣氛接觸的各結構域的交替垂直取向片層的層狀結構域圖案。墊層材料可以是對給定sa材料的結構域具有合適的中性潤濕性質的任何材料。
sa層具有下列特征。sa層包含含有脂族聚碳酸酯嵌段的高chibcp。另外,bcp具有有利于在自組裝過程中形成層狀結構域或圓柱形結構域的結構。也就是說,嵌段共聚物的嵌段的體積分數在有利于形成層狀結構域或圓柱形結構域的范圍內。sa層也布置在對自組裝bcp的結構域中性潤濕的墊層表面上。最后,sa層具有與氣氛接觸的頂表面。氣氛界面,通常是空氣,對bcp非中性,意味著當sa層本質上由bcp構成時,自組裝sa層的頂表面的并非所有結構域與氣氛接觸。在這些條件下,自組裝bcp通常僅一個結構域與氣氛接觸(片層具有平行取向)。一般而言,bcp的chi參數越高,墊層表面和氣氛之間的表面性質失配越大,造成層狀結構域的平行取向。
考慮到sa層的上述特征,由于并非所有結構域能夠“潤濕”氣氛,不含sap的sa層自組裝形成平行取向的層狀結構域。平行取向的層狀結構域在原子力顯微術(afm)高度圖像(下文的示意2)中以島和孔外觀為特征。合意的是包含sa材料和sap的sa層的自組裝形成層狀結構域圖案,其中各結構域與墊層和氣氛接觸。墊層材料可以是對給定sa材料的結構域具有合適的中性潤濕性質的任何材料。
下面的論述集中于通過二嵌段共聚物的自組裝形成的層狀結構域圖案,但也適用于其它嵌段共聚物(例如三嵌段共聚物)和其它結構域狀態(例如圓柱形結構域)。應該理解的是,層圖不是按比例繪制的,也不對可使用下文所述方法制成的可能結構不構成限制。附圖意在用于示例性說明。
不受制于理論,示意1是自組裝二嵌段共聚物的平行取向的層狀結構域的示意圖。各層狀結構域的主平面平行于墊層表面的平面。如圖1a中所示的示意1顯示二嵌段共聚物的嵌段a和b在二嵌段共聚物在對含嵌段a的結構域優先的墊層表面上自組裝后的排列。在這一實例中,墊層和氣氛界面對含嵌段a的結構域具有優先親和性。第一層狀結構域包含嵌段a且第二層狀結構域包含嵌段b。結構域的體相周期性lo由1.0lo(1.0乘以lo)標出。還標出了各二嵌段共聚物大分子、結構域邊界和0.5lo。應該理解的是,在給定層狀結構域(例如示意1的第二層狀結構域)內,來自不同聚合物大分子的嵌段(例如b嵌段)可以首尾相接排列(已顯示)和/或交織(未示出)。各嵌段可具有剛性、非剛性或中間剛性的骨架。各嵌段可具有任何合適的盤繞、旋轉和/或彎曲能力。
對本發明而言,氣氛界面始終對高chibcps是非中性界面。與氣氛界面接觸的本質上由高chibcp構成的sa層幾乎肯定發生自組裝以形成島和孔,其邊界代表平行取向片層中的中斷。這無論墊層對bcp是中性還是非中性都會發生。
示意2是當氣氛對bcp非中性且自組裝過程在中性墊層表面上形成島和孔時自組裝bcp的平行取向的層狀結構域的示意圖。在如圖1b中所示的示意2中,墊層表面與含嵌段a的結構域和含嵌段b的結構域接觸,且只有嵌段a結構域與氣氛接觸。墊層表面的中性(非優先)潤濕性質在平行取向的層狀結構域中造成中斷,以致形成具有大約0.5lo的通過afm得出的階高h'的島和孔。應該理解的是,嵌段a相分離以保持與氣氛的接觸,包括在層狀結構域的中斷區(孔和島的邊界)中。沒有嘗試表征嵌段b在示意2的中斷區中的排列。
下列實施例表明,當sa層的頂表面與氣氛接觸時,包含bcp和所公開的sap的sa層可以自組裝以形成垂直取向的層狀結構域。sap與聚碳酸酯結構域(即包含自組裝bcp的聚碳酸酯嵌段的結構域)優先溶混并與自組裝嵌段共聚物的非聚碳酸酯結構域(例如由乙烯基可聚合單體,如苯乙烯和/或取代苯乙烯制成的嵌段)基本不溶混。如下述實施例中所示,使用sap形成的墊層對bcp的聚碳酸酯結構域具有優先親和性。(這不是sap的預期用途,但可用于證實sap的表面性質)。
不受制于理論,但相信自組裝產生具有比非聚碳酸酯結構域高的sap濃度的聚碳酸酯結構域。在自組裝后聚碳酸酯結構域的較高sap濃度可使得富集sap的聚碳酸酯結構域能夠潤濕氣氛界面,由此允許層狀結構域垂直取向。與氣氛接觸的非聚碳酸酯結構域可以在氣氛界面處實質上不含sap。當嵌段共聚物結構有利于形成圓柱形結構域時,sap的存在可允許形成垂直取向的圓柱形結構域。
示意3是自組裝bcp的垂直取向的層狀結構域的示意圖,其中sa層包含優先可溶于含嵌段b的結構域的sap。sap在嵌段b和嵌段a結構域內由正斜杠標記表示。嵌段b結構域可包含sap的濃度梯度,其中sap濃度在氣氛界面處(未示出)最高。嵌段a結構域可以在氣氛界面處實質上不含sap。在如圖1c中所示的示意3中,片層的主平面垂直于墊層表面的平面以及sa層的主平面取向。各結構域的片層與氣氛和墊層表面接觸。標出了體相周期性lo,以及0.5lo。在這一實例中,墊層表面與自組裝二嵌段共聚物的嵌段a和嵌段b接觸。sap的存在使得含sap的嵌段b結構域能夠接觸氣氛界面。
在下述實施例中,如果包含自組裝bcp的sa層通過原子力顯微術表現出島和/或孔或平行圓柱體,則氣氛界面(空氣界面)對自組裝sa層非中性(不合意)。如果包含自組裝bcp的sa層表現出垂直取向的片層或圓柱體,氣氛界面(空氣界面)對自組裝sa層中性(合意)。
下述實施例表明sap能夠運行形成垂直取向的結構域圖案而無需頂涂層(即在sa層和氣氛界面之間的層)或形貌預圖案來引導高chibcp的自組裝。盡管形貌預圖案對取向控制而言不是必需的,但形貌預圖案可以為其它用途存在。這也在下述實施例中證實。
層狀結構域圖案可具有大約4納米至大約80納米的體相周期性lo,這有助于制造具有大約2納米至大約40納米,更特別大約2納米至大約20納米的半節距的線形態部(features)。
表面活性聚合物(saps)
sap是相選擇性的,意味著sap在通過自組裝形成的聚碳酸酯結構域中具有優先可溶性。因此,聚碳酸酯結構域在自組裝后具有比其它結構域高的sap濃度。自組裝后的聚碳酸酯結構域的sap濃度充分降低聚碳酸酯結構域的表面能以允許聚碳酸酯結構域潤濕氣氛界面并提供各結構域的垂直取向。可以在sa層上不形成sap單層的情況下實現垂直取向。因此,sap可以以sa層的干固體總重量的大于0重量%至大約10重量%,更尤其大約0.1重量%至大約10重量%,再更尤其0.1重量%至大約5重量%的量使用。在一個實施方案中,用于制備sa層的制劑包含基于sa層的干固體總重量計大約0.5重量%至大約5重量%sap。
sap優選是在聚碳酸酯結構域中具有優先可溶性的均聚物或無規共聚物。不排除其它聚合物構造(例如樹枝狀聚合物、星形聚合物和嵌段聚合物),只要sap優先可溶于聚碳酸酯結構域并且在自組裝過程中不形成與聚碳酸酯結構域分開且不同的離散結構域。無規共聚物是包含不同的構成聚合物骨架的重復單元的無規分布的聚合物。無規共聚物名稱可包括分開用于制備聚合物的縮寫單體名稱的"-r-"或"-無規-"。
sap可通過任何合適的聚合技術制備。示例性的聚合技術包括自由基聚合、原子轉移自由基聚合(atrp)、可逆加成-斷裂鏈轉移聚合(raft)、陰離子聚合、開環聚合和開環易位聚合(romp)。用于形成sap的非限制性單體包括乙烯基可聚合單體(例如苯乙烯、丙烯酸酯、甲基丙烯酸酯)和環狀單體(例如環醚、環狀碳酸酯)。
sap包含氟-醇重復單元(hfa重復單元),其包含至少一個如下結構的六氟異丙基醇基團(hfa基團):
因此,saps在本文中也被稱為“氟-醇添加劑”或簡稱為“聚合物添加劑”。在此,星號鍵代表連接點,而非甲基。
氟-醇重復單元可具有根據式(h-1)的結構:
其中
a'是1或2,
q″是具有a'+1的化合價的連接基,且q″包含至少一個碳,且
r″是選自*-h、甲基(*-ch3)、乙基(*-et)和三氟甲基(*-cf3)的一價基團。
q″可包含任何合適的官能團。非限制性的示例性q″基團包括非芳族環狀基團、芳基、烷基取代的芳基、氟取代的芳基、酯基團和酰胺基團。
更具體的q″基團包括示意4中的那些,其中q″的碳1的星號鍵連接到帶有式(h-1)的r″基團的碳1上,且q″的各剩余星號鍵連接到hfa基團上。
示意4.
非限制性的示例性hfa重復單元包括示意5的那些。
示意5.
sap可包含單獨或組合的hfa重復單元。非限制性的示例性含hfa乙烯基可聚合單體(hfa單體)包括示意6的那些。
示意6.
sap可進一步包含一個或多個具有任何合適結構的稀釋劑重復單元。作為非限制性實例,sap可包含一個或多個示意7的二價重復單元,其可通過相應苯乙烯單體的乙烯基聚合形成。
示意7.
當給定基團的星號鍵跨環的鍵時,如上示稀釋劑重復單元中所示,基團可作為該環的任一可能位置異構體或作為該環的位置異構體的混合物存在。下面也遵照這一約定。
用于sap的其它非限制性稀釋劑重復單元包括示意7a的二價氟化酯重復單元,其可通過相應(甲基)丙烯酸酯單體的乙烯基聚合形成。
示意7a.
在一個實施方案中,sap的稀釋劑重復單元包含選自苯基、包含一個或多個氟的苯基、包含一個或多個氟的酯基團、包含一個或多個氟的醚基團和包含一個或多個氟的烷基的官能團。在一個實施方案中,sap包含含五氟苯基的稀釋劑重復單元。在另一實施方案中,sap是包含氟-醇重復單元和一個或多個稀釋劑重復單元的無規共聚物,所述稀釋劑重復單元選自
其中各r″獨立地選自*-h、*-me、*-et和*-cf3,且各n'是1至12的獨立整數。
當存在時,稀釋劑重復單元可以單獨或組合使用。
sap可具有任何合適的端基官能,例如氫、鹵素基團(例如氟、氯、溴、碘)、烷基、烷氧基、酯基團、芳基、非芳族環狀基團、包含上述官能的組合的基團和被一個或多個氟基取代的任何上述基團。
在一個實施方案中,sap是式(h-2)的聚合物:
e'-p'-e″(h-2),
其中
e'是第一端基,
e″是第二端基,
e'和/或e″包含1至25個氟,且
p'是包含至少一個式(h-1)的氟-醇重復單元(hfa重復單元)的聚合物鏈:
其中
a'是1或2,
q″是具有a'+1的化合價的連接基,且q″包含至少一個碳,且
r″是選自*-h、甲基(*-ch3)、乙基(*-et)和三氟甲基(*-cf3)的一價基團。
在另一實施方案中,式(h-2)的e'和/或e″包含10至25個氟。
在另一實施方案中,sap端基e'和/或e″包含含有1至25個氟的酯基團。氟化酯基團的非限制性實例包括示意8的那些。
示意8.
在一個具體實施方案中,sap聚合物包含含有上述氟化酯基團的第一端基e'和為溴的第二端基e″。作為一個實例,下列示意9示出了pf-oibr的atrp聚合形成表面活性聚合物sap7-c。
示意9.
sap可包含基于用于形成sap的重復單元的總摩爾數計20摩爾%至100%,優選40摩爾%至100摩爾%的量的氟-醇重復單元。
sap可具有大約1000至大約50000,更特別1000至大約25000的數均分子量(mn)。sap可具有大約1.0至大約3.0的多分散性指數。
用于取向控制的無規共聚物(墊層)
墊層聚合物優選是能夠發生反應以與基底的另一層形成共價鍵的無規共聚物。更具體的墊層聚合物包含含有乙烯類骨架部分和含聚碳酸酯鏈的側鏈部分的重復單元。側鏈的聚碳酸酯鏈可包含1個或更多個碳酸酯重復單元,優選2至大約40個碳酸酯重復單元。在一個實施方案中,墊層聚合物的聚碳酸酯側鏈的碳酸酯重復單元具有與sa層中所用的bcp的聚碳酸酯嵌段的碳酸酯重復單元相同的化學結構(下文進一步描述)。包含聚碳酸酯側鏈的墊層聚合物可通過環狀碳酸酯的使用具有一個或多個能夠引發rop的側羥基的聚合物大分子引發劑的開環聚合(rop)制備。聚合物大分子引發劑可通過一種或多種包含醇側基(例如甲基丙烯酸羥乙酯,hema)或經保護的醇基團(其隨后脫保護)的乙烯基可聚合單體的無規聚合制備。
更具體地,用于墊層的無規共聚物包含式(a-1)的第一重復單元:
其中
rx是選自h、甲基、乙基和三氟甲基(*-cf3)的一價基團,且
rb是包含芳環的一價基團。
應該理解的是,式(a-1)的兩個星號鍵代表與聚合物鏈的其它重復單元或聚合物鏈端基的連接點,碳1和2是聚合物骨架的乙烯碳,rx是連接到聚合物骨架的碳1上的第一側鏈,且rb是連接到聚合物骨架的碳1上的第二側鏈。
式(a-1)的非限制性rb基團包括取代和未取代芳基。示例性rb基團列在下示示意10中,其中羰基或芳環的星號鍵連接到式(a-1)的標作1的碳上。
示意10.
在一個實施方案中,式(a-1)的rx是氫,且rb是苯基。
無規共聚物包含式(a-2)的第二重復單元:
其中
rx是選自h、甲基、乙基和三氟甲基(*-cf3)的一價基團,且
z'是包含兩個或更多個獨立地選自酯基團、碳酸酯基團、酰胺基團、氨基甲酸酯基團及其組合的含羰基官能團的一價基團。
應該理解的是,rx是連接到式(a-2)的聚合物骨架碳1上的第二重復單元的第一側鏈,且z'是連接到式(a-2)的聚合物骨架碳1上的第二重復單元的第二側鏈。
式(a-2)的第二重復單元包括獲自苯乙烯、丙烯酸酯、甲基丙烯酸酯、丙烯酰胺、甲基丙烯酰胺等的那些,其在乙烯基聚合之前或之后改性以包含所述兩個或更多個含羰基官能團。
式(a-2)的更具體的z'基團包括式(a-2a)的那些:
其中
l'是包含2至10個碳的二價連接基,
qa是*-o-*或*-n(h)-*,
qb是*-o-*或*-n(h)-*,且
w'是包含至少一個碳的基團。
式(a-2a)的星號鍵連接到式(a-2)的碳1上。
式(a-2a)的z'基團的非限制性實例包括示意11的那些。示意11.
在一個實施方案中,z'是
式(a-2)的其它更具體的z'基團包括式(a-2b)的那些:
其中
a'代表重復單元的平均數并具有1或更大的值,
e1是獨立地選自氫和包含1至10個碳的基團的一價端基,
各j'是選自氫和包含1至10個碳的基團的獨立一價基團,
l'是包含2至10個碳的二價連接基,
qa是*-o-*或*-n(h)-*,
qb是*-o-*或*-n(h)-*,且
各ra是選自氫、甲基和乙基的獨立一價基團。
式(a-2b)的星號鍵連接到式(a-2)的碳1上。z'聚合物的平均聚合度a'優選為1至大約40,更優選1至大約20。ra是z'聚合物的第一側鏈,且j'是z'聚合物的第二側鏈(上文標記)。
式(a-2b)的z'基團的非限制性實例包括示意12的那些。
示意12.
在一個實施方案中,qa和qb是*-o-*,l'是亞乙基(*-ch2ch2-*),ra是氫,且j'是式(a-2b)的氫。在另一實施方案中,z'是
其中a'代表重復單元的平均數并具有1或更大的值。
式(a-2)的另一些更具體的z'基團包括式(a-2c)的那些:
其中
a'代表括號中圍住的重復單元的平均數并具有1或更大的值,
u'是0或1,
e1是獨立地選自氫和包含1至10個碳的基團的一價端基,
各j'是選自氫和包含1至10個碳的基團的獨立一價基團,
la是包含1至5個碳的二價連接基,
各ra是選自氫、甲基和乙基的獨立一價基團,且
當a'是1時,la、ra、j'和/或e'包含選自酯、碳酸酯、氨基甲酸酯及其組合的含羰基官能團。
式(a-2c)的星號鍵連接到式(a-2)的碳1上。z'聚合物的平均聚合度a'優選為1至大約40,更優選1至大約20。ra是z'聚合物的第一側鏈,且j'是z'聚合物的第二側鏈。
式(a-2c)的z'基團的非限制性實例包括示意13的那些,其中a'具有1至大約40的平均值,且rf是包含1至10個碳的基團(例如甲基、乙基、丙基、丁基、芐基)。
示意13.
式(a-2)的第二重復單元的非限制性實例包括示意13b的那些,其中a'具有1至大約40的平均值,且rf是包含1至10個碳的基團。
示意13b.
在一個實施方案中,墊層無規共聚物的第二重復單元選自
其中a'代表聚合度并具有大約1至大約40的平均值且rf是甲基、乙基、丙基、苯基和芐基的一員。
用于墊層的無規共聚物優選包含24:76至88:12的第一重復單元:第二重復單元摩爾比。
用于墊層的無規共聚物進一步包含式(a-3)的第三重復單元:
其中
rz是選自h、甲基、乙基和三氟甲基(*-cf3)的一價基團,
l″是包含1至10個碳的獨立二價連接基,且
k'是能與親核體反應以形成共價鍵的一價親電子基團。
式(a-3)的非限制性l″基團包括酯、酰胺、芳基、芳基酯和芳基酰胺基團。示例性l″基團包括下列示意14中所列的那些,其中羰基或芳環的星號鍵(即各結構中的最左星號鍵)連接到式(a-3)的聚合物骨架碳1上。
示意14.
k'可包含選自活性羧酸酯基團(例如對硝基苯基酯、五氟苯基酯)、鹵素基團(例如氯、溴和碘)、磺酸酯(例如對甲苯磺酸酯、甲磺酸酯)、含環氧基的基團等的親電子基團。在一個實施方案中,k'包含環氧基。
更具體的l″-k'基團包括示意15的那些,其中來自羰基或芳環的星號鍵連接到式(a-3)的標作1的碳上。
示意15.
在一個實施方案中,rz是式(a-3)的甲基且式(a-3)的l″-k'是
用于墊層的更具體無規共聚物具有根據式(a-4)的結構:
其中
各下標x'、y'和z'代表各自的括號中圍住的重復單元的平均數并獨立地具有大于1的平均值,
e'和e″是獨立地選自氫、鹵素和包含1-10個碳的基團的一價端基,
各k'是能與基底表面反應以形成共價鍵的一價基團,
l'是包含2至10個碳的二價連接基,
各l″是包含1至10個碳的獨立二價連接基,
qa是*-o-*或*-n(h)-*,
qb是*-o-*或*-n(h)-*,
各rb是包含1個或更多個芳環的獨立一價基團,
rx、ry和rz各自是獨立地選自氫、甲基、乙基和三氟甲基(*-cf3)的一價基團,且
w'是包含至少一個碳的基團。
在一個實施方案中,各rx是氫,各rb是苯基,qa是*-o-*且qb是*-o-*。在另一實施方案中,l'是亞乙基(*-ch2ch2-*)且w'是甲基。
在此,式a-4也可以寫作式(a-5):
方括號內的重復單元的垂直堆疊表示聚合物鏈中的重復單元的無規分布。在重復單元的無規分布中,其星號鍵與左方括號搭接的給定重復單元可連接到其星號鍵與右方括號搭接的不同重復單元的原子中心或端基e'上。同樣地,其星號鍵與右方括號搭接的給定重復單元可連接到其星號鍵與左方括號搭接的不同重復單元的原子中心或端基e″上。端基e'可連接到具有與左方括號搭接的星號鍵的任一重復單元上。端基e″可連接到具有與右方括號搭接的星號鍵的任一重復單元上。
用于墊層的另一些更具體的無規共聚物具有根據式(a-6)的結構:
其中
各下標a'、x'、y'和z'代表各自的括號中圍住的重復單元的平均數并獨立地具有大于1的平均值,
e'和e″是獨立地選自氫、鹵素和包含1-10個碳的基團的一價端基,
e1是獨立地選自氫和包含1至10個碳的基團的一價端基,
各j'是選自氫和包含1至10個碳的基團的獨立一價基團,
各k'是能與基底表面反應以形成共價鍵的一價基團,
各l'是包含2至10個碳的二價連接基,
各l″是包含1至10個碳的獨立二價連接基,
qa是*-o-*或*-n(h)-*,
qb是*-o-*或*-n(h)-*,
各ra是選自氫、甲基和乙基的獨立一價基團,
各rb是包含1個或更多個芳環的獨立一價基團,且
rx、ry和rz各自是獨立地選自氫、甲基、乙基和三氟甲基(*-cf3)的一價基團。
在一個實施方案中,各rx是氫,各rb是苯基,qa是*-o-*且qb是*-o-*,ra是氫,且j'是氫。在另一實施方案中,l'是亞乙基(*-ch2ch2-*)且e1是乙酰基。
式a-6也可以寫作式(a-7):
用于墊層的另一些更具體的無規共聚物具有根據式(a-8)的結構:
其中
x'、y'和z'各自代表各自的括號中圍住的各重復單元的平均數并具有大于1的平均值,
a'代表括號中圍住的各重復單元數并具有1或更大的平均值,
e'和e″是獨立地選自氫、鹵素和包含1-10個碳的基團的一價端基,且
e1是獨立地選自氫和包含1-10個碳的基團的一價端基。
優選地,x'是20至80,y'是1至20,z'是1至5,且a'是1至40。在一個實施方案中,e1是乙酰基。在另一實施方案中,a'是大約1至大約10。
式(a-8)的結構也可以由式(a-9)表示:
用于取向控制層的無規共聚物的制備
用于墊層的無規共聚物可通過包含含芳環的乙烯基可聚合單體、含聚碳酸酯或聚酯碳酸酯側鏈的第二乙烯基可聚合單體和含能與親核體反應以產生共價鍵的親電子基團的第三乙烯基可聚合單體的混合物的共聚制備。示意16中示出了該方法,其中使用單體苯乙烯(sty)、hema-ptmc和甲基丙烯酸縮水甘油酯(gma)。用于乙烯基聚合的催化劑包括自由基引發劑(例如偶氮二異丁腈(aibn))。
示意16.
hema-ptmc可如下面示意17中所示通過使用甲基丙烯酸羥乙酯(hema)作為聚合引發劑的碳酸三亞甲酯(tmc)的有機催化開環聚合(rop)和用乙酰氯將所得聚碳酸酯封端制備。用于rop的催化劑包括有機堿(例如1,8-二氮雜雙環[5,4,0]十一碳-7-烯(dbu))和磷酸酯(例如磷酸二苯酯(dpp))。
示意17.
制備用于墊層的無規共聚物的第二種方法包括如示意18中所示由前體無規共聚物的側鏈上的親核位點通過開環聚合生長聚碳酸酯或聚酯碳酸酯鏈。
示意18.
在這一實例中,苯乙烯(sty)、甲基丙烯酸羥乙酯(hema)和甲基丙烯酸縮水甘油酯(gma)的無規共聚物,被稱作p(sty-hema-gma),是用于tmc的開環聚合的大分子引發劑。大分子引發劑p(sty-hema-gma)的各親核羥基充當tmc的rop的潛在引發位點。所得聚合物是包含多個帶有聚碳酸酯鏈的側鏈的無規接枝共聚物。
用于自組裝的嵌段共聚物
嵌段共聚物名稱可包括"-b-"或"-嵌-",分隔用于制備該聚合物的單體縮寫名。用于自組裝的嵌段共聚物可包含兩個或更兩個嵌段,優選2至4個嵌段。至少一個嵌段包含碳酸酯重復單元。例如,嵌段共聚物可包含通過連接基連接的一個聚苯乙烯(ps)嵌段和一個聚碳酸酯(pc)嵌段。作為另一實例,嵌段共聚物可包含通過一個或兩個線性聚合物鏈(即不是大環)形式的連接基連接的兩個ps嵌段和一個pc嵌段。作為另一實例,嵌段共聚物可包含通過1至3個連接基連接的兩個ps嵌段和兩個pc嵌段。
下面討論涉及二嵌段聚合物(a-b),但也可適用于三嵌段聚合物和其它嵌段聚合物構造(例如包含連接到多價核上的3個或更多個聚合物臂的星形聚合物、雜臂(mikto-arm)星形聚合物,其中一個或多個臂包含與其它臂不同的重復單元)。
第一嵌段(嵌段a)包含含重復官能化乙烯單元的骨架(例如如聚苯乙烯骨架中)。第二嵌段(嵌段b)包含至少一個脂族碳酸酯重復單元(即在聚合物骨架中包含脂族碳酸酯基團)。選擇嵌段以基本彼此不溶混。另外,優選的是嵌段聚合物的第一嵌段和第二嵌段相對于用于沉淀嵌段聚合物的溶劑混合物具有下列溶解性質:i)第一嵌段和第二嵌段基本不溶于溶劑混合物的第一溶劑,ii)第一嵌段基本不溶于溶劑混合物的第二溶劑,和iii)第二嵌段可溶于溶劑混合物的第二溶劑。也就是說,第一溶劑是第一嵌段和第二嵌段的非溶劑,第二溶劑是第一嵌段的非溶劑,且第二溶劑是第二嵌段的溶劑。
通過嵌段聚合物的自組裝形成的特定體相結構單元取決于第一嵌段與第二嵌段的體積比。給定嵌段的體積是指嵌段占據的體積,其取決于嵌段的分子質量。例如,當第一嵌段a與第二嵌段b的體積比大于大約80:20時,嵌段聚合物可形成在由第一嵌段構成的基質中的由第二嵌段構成的球體的有序陣列。當第一嵌段與第二嵌段的體積比在大于大約80:20的范圍內時,嵌段共聚物可形成在由第一嵌段構成的基質中的由第二嵌段的球體的有序陣列。當第一嵌段與第二嵌段的體積比在大約80:20至大約60:40的范圍內時,嵌段聚合物可形成在由第一嵌段構成的基質中的由第二嵌段構成的圓柱體的有序陣列。當第一嵌段與第二嵌段的體積比小于大約60:40至大約40:60時,嵌段聚合物可形成交替片層(即與由第二嵌段構成的結構域交替的由第一嵌段構成的結構域的陣列)。作為一個實例,包含20體積%或更少的聚苯乙烯(ps)嵌段的聚苯乙烯-b-聚甲基丙烯酸甲酯嵌段共聚物(ps-b-pmma)可以自組裝以形成在聚甲基丙烯酸甲酯(pmma)基質中的ps球體。作為另一實例,包含大約20體積%至40體積%ps的ps-b-pmma嵌段共聚物可以自組裝以形成在pmma基質中的ps圓柱體。可通過控制各嵌段的平均分子量調節第一嵌段與第二嵌段之間的體積比。
更具體地,基于嵌段聚合物大分子的平均總體積,第一嵌段與第二嵌段的體積比可以為大約15:85至大約85:15。優選地,為了形成交替片層,第一嵌段與第二嵌段的體積比可以為大約40:60至大約60:40,更優選45:55至55:45,,最優選48:52至52:48。為了形成圓柱體,第一嵌段與第二嵌段的體積比可以為大約74:26至大約63:37,更優選大約72:28至大約65:35。
可以使用已知的干和/或濕蝕刻技術相對于另一嵌段(例如通過蝕刻技術)選擇性除去嵌段聚合物的嵌段之一以形成由剩余嵌段構成的結構形態部。結構形態部可具有任何合適的形式,例如線圖案、孔圖案和/或其它圖案。
rop聚合物引發劑優選是乙烯基聚合的產物。乙烯基可聚合單體包括苯乙烯、丙烯酸酯、甲基丙烯酸酯、它們的取代衍生物等。乙烯基可聚合單體可以單獨或組合使用以使用任何合適的聚合技術形成rop聚合物引發劑,包括但不限于自由基聚合、陰離子聚合、陽離子聚合、原子轉移自由基聚合(atrp)、氮氧介導聚合(nmp)和/或可逆加成-斷裂鏈轉移(raft)聚合。
更具體地,嵌段共聚物的第一嵌段包含式(b-1)的重復單元:
其中
rw是選自h、甲基、乙基和三氟甲基(*-cf3)的一價基團,且
rd是包含連接到碳1上的芳環的一價基團。
式(b-1)的非限制性rd基團包括取代和未取代的芳基。示例性的rd基團列在下面示意19中,其中羰基或芳環的星號鍵連接到式(b-1)的標作1的碳上。
示意19.
在一個實施方案中,式(b-1)的rw是氫,且rd是苯基。式(b-1)的重復單元可以單獨或組合存在。
第一嵌段可以是式(b-1)的重復單元的均聚物或包含式(b-1)的重復單元和/或第二重復單元的組合的無規共聚物鏈。
二嵌段共聚物的第二嵌段包含至少一個式(b-2)的脂族碳酸酯重復單元:
其中
re是選自氫、甲基和乙基的獨立一價基團,且
j″是選自氫和包含1至20個碳的基團的獨立一價基團。
用于自組裝和定向自組裝的更具體的嵌段共聚物具有根據式(b-3)的結構,其中方括號代表嵌段共聚物的單獨嵌段a和b:
其中
各下標m'和n'代表括號中圍住的各重復單元的平均數,并獨立地具有大于1的平均值,
e2是選自氫和包含1-10個碳的基團的一價端基,
g'是選自氫、鹵素和包含1-10個碳的基團的一價端基,
g″是包含1-20個碳的二價連接基,
各j″是選自氫和包含1至20個碳的基團的獨立一價基團,
各rd是包含芳環的獨立一價基團,
各rw是選自h、甲基、乙基和三氟甲基(*-cf3)的獨立一價基團,
各re是選自氫、甲基和乙基的獨立一價基團,且
各x″是選自*-o-*、*-s-*、*-n(h)-*和*-n(r″)-*的獨立二價基團,其中r″是包含1至6個碳的一價基團。
在一個實施方案中,各j″是氫,各re是氫,各rw是氫,各rd是苯基,e2是乙酰基,且x″是*-o-*。
用于自組裝的嵌段共聚物的更具體的實例具有根據式(b-4)的結構:
其中
各下標m'和n'代表括號中圍住的各重復單元的平均數,并獨立地具有大于1的平均值,
各re是選自氫、甲基和乙基的獨立一價基團,且
各j″是選自氫和包含1至20個碳的基團的獨立一價基團。
在一個實施方案中,各re是氫且各j″是氫。在另一實施方案中,各re是甲基且各j″是*-co2et。
用于自組裝的嵌段共聚物的制備
用于自組裝的嵌段共聚物優選通過使用具有衍生自乙烯基可聚合單體的骨架的聚合物引發劑的環狀碳酸酯單體的開環聚合制備。為了形成二嵌段和三嵌段共聚物,聚合物引發劑可分別包含1和2個能夠引發環狀羰基單體的rop的親核基團(例如*-oh、*-nh2)。rop反應混合物包含環狀羰基單體、rop催化劑、溶劑和聚合物引發劑。rop催化劑優選是酸催化劑(例如磷酸二苯酯)。
下列形成二嵌段共聚物的方法可用于制備三嵌段聚合物和其它嵌段聚合物構造。所述方法提供實質上不含任何聚碳酸酯均聚物和/或聚碳酸酯無規共聚物的嵌段共聚物。
方法1
這種方法使用溶劑混合物在rop進行至一定持續時間時分離形成的初始嵌段聚合物,所述時間對應于環狀碳酸酯單體消耗為大約50%至100%,更特別大約85%至100%。對于給定的一組反應條件(例如溫度、溶劑、氣氛類型、相對摩爾量和其它反應參數),可以使用任何合適的分析技術(例如質子核磁共振(1hnmr))監測環狀碳酸酯單體的消耗。
rop產生含活性端基(其是能夠發生進一步鏈生長和/或引發不同環狀羰基單體的rop的親核基團)的初始嵌段聚合物。優選地,活性端基通過將封端劑添加到反應混合物上鈍化,由此終止聚合并形成含有受保護的親核端基的封端初始嵌段聚合物。封端初始嵌段聚合物不能引發rop。作為一個實例,通過環狀碳酸酯單體的rop形成的聚碳酸酯具有含親核羥基的活性末端,其可通過添加合適的酰化劑(例如乙酸酐)鈍化以形成受保護的羥基(例如作為乙酰基酯)。
分離出的初始嵌段聚合物或封端初始嵌段聚合物(“粗制嵌段聚合物”)可含有未共價連接到聚合物引發劑上的衍生自環狀羰基單體的聚合物雜質。聚合物雜質可包括通過痕量水引發的聚碳酸酯均聚物和/或通過初始嵌段聚合物的聚碳酸酯骨架上的活性羥端基的回咬(backbiting)形成的環狀聚碳酸酯。這些雜質可不利地影響初始嵌段聚合物的自組裝性質。
可以通過下列分離法除去聚合物雜質。制備第一溶液,其含有溶解在最少量的能夠溶解嵌段聚合物的各嵌段的溶劑(例如thf)中的初始嵌段聚合物。第一溶液含有基于第一溶液的總重量計大約20重量%濃度的初始嵌段聚合物。然后將第一溶液添加到過量(按重量計粗制聚合物的量的大約200至400倍)的包含大約40:60至大約60:40體積比的第一溶劑和第二溶劑的溶劑混合物中,其中第一溶劑是第一嵌段和第二嵌段的非溶劑,第二溶劑是第一嵌段的非溶劑和第二嵌段的溶劑。在一個實施方案中,第一溶劑是meoh且第二溶劑是乙腈。溶劑混合物選擇性溶解聚合物雜質,以使最終嵌段聚合物作為可基本不含聚合物雜質的固體沉淀出來。分離程序視需要可以重復一次或多次以形成用于自組裝用途的嵌段聚合物。
方法2
在第二種方法中,使用包括聚合物引發劑的給定反應條件組進行試驗rop。如方法1中那樣隨rop持續時間t監測消耗的環狀羰基單體的量(例如,消耗%),允許rop進行至環狀羰基單體消耗85%至100%。繪制環狀碳酸酯的消耗%vs.以分鐘計的rop持續時間t的曲線圖。
由所收集的數據點的散布圖,可以將二階多項式函數f(t)(即趨勢線)擬合至繪制的點,其中f(t)表示消耗的環狀羰基單體的量作為rop持續時間t的函數。f(t)的r2(平方r)系數優選具有大約0.85至1.0,更優選0.9至1.0的值。
使用f(t)的表達式,可以計算與環狀羰基單體的50%消耗對應的時間t1。
然后對各測量時間t計算f(t)的一階導數,標作f'(t)。
然后測定在50%環狀單體轉化率下的f'(t)值。使用在50%環狀單體轉化率下的f'(t1)值,確定與-10%和-20%的斜率變化對應的rop持續時間t2和t3,所述變化是相對于在50%環狀羰基單體消耗下的斜率。
然后使用給定反應條件進行rop,在持續時間(t')停止rop,其中t1≤t'≤t3,更優選t2≤t'≤t3。使用這些改良反應條件,可以直接獲得不含或基本不含不包括衍生自聚合物引發劑的嵌段的聚合物雜質的用于自組裝的嵌段聚合物。任選地,嵌段聚合物可以進一步用如上文在方法1下描述的溶劑混合物處理以除去任何剩余聚合物雜質。
方法3
在方法3中,如上文在方法2下所述獲得f'(t)的數學表達式。然后對各rop持續時間t計算f'(t)的值。使用f'(t)的值,對大于0的各rop持續時間計算相鄰rop持續時間之間的f'(t)變化。例如,在持續時間tn下的f'(t)變化,標作δf'(tn),等于f'(tn)-f'(tn-1),其中n是正整數且tn>0。
對具有倒拋物線形狀的δf'(t)vs.t的散布圖獲得二階多項式趨勢線d(t)。d(t)具有一階導數d'(t),其在一定rop持續時間t″>0下等于0,所述t″小于對應于100%環狀羰基單體消耗的持續時間。
使用給定反應條件重復rop,在0.8t″至大約t″終止rop。所得最終嵌段聚合物可以不含或基本不含不包括衍生自聚合物引發劑的嵌段的聚合物雜質。任選地,嵌段聚合物可以進一步用如上文在方法1下描述的溶劑混合物處理以除去存在的任何聚合物雜質。
環狀羰基單體
示例性的環狀羰基單體包括示意20的環狀碳酸酯化合物,其可用于例如形成初始嵌段聚合物的聚碳酸酯嵌段。
示意20.
其它環狀羰基單體包括環酯(內酯),如示意21的化合物。
示意21.
上述環狀羰基單體可通過從溶劑如乙酸乙酯中重結晶或通過其它已知提純方法提純,特別注意從單體中除去盡可能多的水。
用于嵌段共聚物的rop引發劑
用于開環聚合的引發劑通常包括親核基團,如醇、伯胺、仲胺和硫醇。在此,用于嵌段共聚物的rop引發劑是包含衍生自可聚合乙烯基單體(苯乙烯、甲基丙烯酸酯、丙烯酸酯、甲基丙烯酰胺、丙烯酰胺、乙烯基酯)的聚合物骨架的聚合物引發劑。一種示例性聚合物引發劑是下示官能化聚苯乙烯引發劑ps-oh。
聚合物引發劑優選包含一個或兩個用于引發rop和分別形成二嵌段、三嵌段和四嵌段共聚物的親核羥基。在一個實施方案中,聚合物引發劑包含兩個親核引發基團,并且通過rop形成的嵌段共聚物是包含4個臂的雜臂星形聚合物。雜臂星形聚合物具有包含連接到星形聚合物的核上的3個或更多個聚合物臂的化學結構,且至少一個臂具有與另一臂不同的聚合物組成。
聚合物引發劑的數均分子量可以為1000至1,000,000,更尤其是1000至100,000,再更尤其是1000至50,000。
形成嵌段共聚物的一種示例性的非限制性反應顯示在示意22中,使用另一大分子引發劑azps-oh。
示意22.
開環聚合(rop)
用于開環聚合的方法、條件和材料的下列描述適用于制備用于取向控制材料的無規共聚物和/或用于自組裝的嵌段聚合物。
開環聚合可以在大約環境溫度或更高的溫度,15℃至100℃,更尤其環境溫度下進行。反應時間隨溶劑、溫度、攪拌速率、壓力和設備而變,但聚合通常在大約1小時至大約48小時內完成。
rop反應可以使用或不使用溶劑,優選使用溶劑進行。示例性的溶劑包括二氯甲烷、氯仿、苯、甲苯、二甲苯、氯苯、二氯苯、三氟甲苯、石油醚、乙腈、戊烷、己烷、庚烷、2,2,4-三甲基戊烷、環己烷、二乙醚、叔丁基甲基醚、二異丙基醚、二氧雜環己烷、四氫呋喃或包含上述溶劑之一的組合。當存在溶劑時,合適的單體濃度為大約0.1至5摩爾/升,更特別是大約0.2至4摩爾/升。
無論在溶液中還是在本體中進行,rop聚合使用惰性(即干燥)氣氛,如氮氣或氬氣,并在100至500mpa(1至5atm)的壓力下,更通常在100至200mpa(1至2atm)的壓力下進行。在反應完成時,可以使用減壓除去溶劑。
rop催化劑
對rop催化劑沒有限制。用于rop聚合的不太優選的催化劑包括金屬氧化物,如四甲氧基鋯、四-異丙氧基鋯、四-異丁氧基鋯、四-正丁氧基鋯、四-叔丁氧基鋯、三乙氧基鋁、三正丙氧基鋁、三-異丙氧基鋁、三正丁氧基鋁、三-異丁氧基鋁、三仲丁氧基鋁、單仲丁氧基-二-異丙氧基鋁、乙酰乙酸乙酯二異丙醇鋁、三(乙酰乙酸乙酯)鋁、四乙氧基鈦、四-異丙氧基鈦、四-正丙氧基鈦、四-正丁氧基鈦、四-仲丁氧基鈦、四-叔丁氧基鈦、三-異丙氧基鎵、三-異丙氧基銻、三-異丁氧基銻、三甲氧基硼、三乙氧基硼、三-異丙氧基硼、三正丙氧基硼、三-異丁氧基硼、三正丁氧基硼、三仲丁氧基硼、三叔丁氧基硼、三-異丙氧基鎵、四甲氧基鍺、四乙氧基鍺、四-異丙氧基鍺、四-正丙氧基鍺、四-異丁氧基鍺、四-正丁氧基鍺、四-仲丁氧基鍺和四-叔丁氧基鍺;鹵化化合物,如五氯化銻、氯化鋅、溴化鋰、氯化錫(iv)、氯化鎘和三氟化硼二乙醚;烷基鋁,如三甲基鋁、三乙基鋁、二乙基氯化鋁、乙基二氯化鋁和三-異丁基鋁;烷基鋅,如二甲基鋅、二乙基鋅和二異丙基鋅;叔胺,如三烯丙基胺、三乙胺、三-正辛基胺和芐基二甲基胺;雜多酸,如磷鎢酸、磷鉬酸、硅鎢酸,以及它們的堿金屬鹽;鋯化合物,如酰氯鋯、辛酸鋯、硬脂酸鋯和硝酸鋯。更特別地,鋯催化劑可以是辛酸鋯、四烷氧基鋯或三烷氧基鋁化合物。
優選的rop催化劑是其化學式不含金屬的有機催化劑。用于環狀羰基單體的rops的堿有機催化劑包括叔胺,如三烯丙基胺、三乙胺、三-正辛基胺和芐基二甲基胺、4-二甲基氨基吡啶、膦、n-雜環卡賓(nhc)、雙官能氨基硫脲、磷腈、脒和胍。
硫脲rop有機催化劑是n-(3,5-三氟甲基)苯基-n’-環己基-硫脲(tu):
另一些rop有機催化劑包含至少一個1,1,1,3,3,3-六氟丙-2-醇-2-基(hfa)基團。單供體氫鍵催化劑具有式(c-1):
r2–c(cf3)2oh(c-1),
其中r2代表氫或具有1至20個碳的一價基團,例如烷基、取代烷基、環烷基、取代環烷基、雜環烷基、取代雜環烷基、芳基、取代芳基或其組合。示例性的單供體氫鍵合催化劑列在示意23中。
示意23.
雙供體氫鍵合催化劑具有兩個hfa基團,如通式(c-2)所示:
其中r3是含有1至20個碳的二價橋連基,如亞烷基、取代亞烷基、亞環烷基、取代亞環烷基、亞雜環烷基、取代亞雜環烷基、亞芳基、取代亞芳基或其組合。代表性的式(c-2)的雙氫鍵合催化劑包括示意24中所列的那些。在一個具體實施方案中,r2是亞芳基或取代亞芳基,且hfa基團占據芳環上的在彼此的間位的位置。
示意24.
優選的氫鍵合催化劑包括4-hfa-st、4-hfa-tol、hftb、nftb、hpip、3,5-hfa-ma、3,5-hfa-st、1,3-hfab、1,4-hfab及其組合。
hfa催化劑可結合到載體上。在一個實施方案中,載體包含聚合物、交聯聚合物珠、無機粒子或金屬粒子。含hfa的聚合物可通過已知方法形成,包括含hfa的單體(例如甲基丙烯酸酯單體3,5-hfa-ma或苯乙烯基單體3,5-hfa-st)的直接聚合。可發生直接聚合(或與共聚單體的聚合)的含hfa的單體中的官能團包括丙烯酸酯、甲基丙烯酸酯、α,α,α-三氟甲基丙烯酸酯、α-鹵代甲基丙烯酸酯、丙烯酰氨基、甲基丙烯酰氨基、降冰片烯、乙烯基、乙烯基醚和本領域中已知的其它基團。連接基的實例包括c1-c12烷基、c1-c12雜烷基、醚基團、硫醚基團、氨基、酯基團、酰胺基團及其組合。還考慮包含帶電含hfa基團的催化劑,所述含hfa基團通過離子締合鍵合到聚合物或載體表面上的帶相反電荷位點上。
最優選地,rop催化劑是酸有機催化劑(例如磷酸二苯酯(dpp)、三氟甲磺酸(triflicacid)等)。
rop反應混合物包含至少一種rop催化劑,和如果適當的話數種rop催化劑一起。rop催化劑相對于環狀羰基單體以1/20至1/40,000摩爾的比例,優選相對于環狀羰基單體以1/1,000至1/20,000摩爾的比例添加。
rop加速劑
rop聚合可以在任選加速劑,特別是氮堿存在下進行。下面列出示例性的氮堿加速劑,包括吡啶(py)、n,n-二甲基氨基環己烷(me2ncy)、4-n,n-二甲基氨基吡啶(dmap)、反式1,2-雙(二甲基氨基)環己烷(tmchd)、1,8-二氮雜雙環[5.4.0]十一碳-7-烯(dbu)、1,5,7-三氮雜雙環[4.4.0]癸-5-烯(tbd)、7-甲基-1,5,7-三氮雜雙環[4.4.0]癸-5-烯(mtbd)、(-)-鷹爪豆堿(sp)、1,3-雙(2-丙基)-4,5-二甲基亞咪唑-2-基(im-1)、1,3-雙(2,4,6-三甲基苯基)亞咪唑-2-基(im-2)、1,3-雙(2,6-二-i-丙基苯基)亞咪唑-2-基(im-3)、1,3-雙(1-金剛烷基)亞咪唑-2-基(im-4)、1,3-二-i-丙基亞咪唑-2-基(im-5)、1,3-二-t-丁基亞咪唑-2-基(im-6)、1,3-雙(2,4,6-三甲基苯基)-4,5-二氫亞咪唑-2-基(im-7)、1,3-雙(2,6-二-i-丙基苯基)-4,5-二氫亞咪唑-2-基、1,3-雙(2,6-二-i-丙基苯基)-4,5-二氫亞咪唑-2-基(im-8)或它們的組合,如示意25中所示。
示意25.
在一個實施方案中,加速劑具有2或3個氮,各自能夠作為路易斯堿參與,例如在結構(-)-鷹爪豆堿中。更強的堿通常改進聚合速率。
催化劑和加速劑可以是相同材料。例如,一些開環聚合可以僅使用1,8-二氮雜雙環[5.4.0]-十一碳-7-烯(dbu)進行,不存在另一催化劑或加速劑。
催化劑優選以環狀羰基單體總摩爾數的大約0.2至20摩爾%、0.5至10摩爾%、1至5摩爾%或1至2.5摩爾%的量存在。
當使用氮堿加速劑,其優選以環狀羰基單體總摩爾數的0.1至5.0摩爾%、0.1至2.5摩爾%、0.1至1.0摩爾%或0.2至0.5摩爾%的量存在。如上所述,在一些情況下,根據特定環狀羰基單體,催化劑和氮堿加速劑可以是相同化合物。
引發劑的量基于二親核引發劑中的每親核引發劑基團的當量分子量計算。引發劑基團優選以環狀羰基單體總摩爾數的0.001至10.0摩爾%、0.1至2.5摩爾%、0.1至1.0摩爾%和0.2至0.5摩爾%的量存在。例如,如果引發劑的分子量為100克/摩爾且引發劑具有2個羥基,每羥基的當量分子量為50克/摩爾。如果聚合需要5摩爾%羥基/摩爾單體,引發劑的量為0.05×50=2.5克/摩爾單體。
在一個具體實施方案中,基于引發劑的每親核引發劑基團的當量分子量計,催化劑以大約0.2至20摩爾%的量存在,氮堿加速劑以0.1至5.0摩爾%的量存在,且引發劑的親核引發劑基團以0.1至5.0摩爾%的量存在。
可以通過選擇性沉淀或在固體負載型催化劑的情況下通過簡單地過濾除去催化劑。嵌段聚合物可包含基于嵌段共聚物和殘留催化劑的總重量計0重量%(重量百分比)或更大量的殘留催化劑。殘留催化劑的量也可以為嵌段共聚物和殘留催化劑的總重量的小于20重量%、小于15重量%、小于10重量%、小于5重量%、小于1重量%或最尤其小于0.5重量%。
封端劑
封端劑可防止進一步的鏈生長并穩定反應性端基以免發生不想要的副反應,如斷鏈。封端劑包括例如用于將端羥基轉化成酯的化合物,如酸酐(例如乙酸酐)、酰氯(乙酰氯)和/或活性酯(例如對硝基苯基酯)。另一些封端劑包括烷基和芳基異氰酸酯,其與端羥基形成氨基甲酸酯(尿烷)。另一些封端劑包括能夠形成烷基醚、芳族醚,包括芐基醚,甲硅烷基醚、縮醛、縮酮等的烷基化劑。再另一些封端劑包括任何上述封端劑的全鹵化(例如全氟化)衍生物。在一個實施方案中,封端劑是乙酸酐,其將反應性羥端基轉化成乙酸酯基團。
平均分子量.
用于自組裝的嵌段聚合物優選具有至少1500克/摩爾,更尤其是1500克/摩爾至1,000,000克/摩爾、4000克/摩爾至150000克/摩爾或4000克/摩爾至50000克/摩爾的如通過尺寸排阻色譜法測定的數均分子量mn。在一個實施方案中,最終嵌段聚合物具有8,000至40,000克/摩爾的數均分子量mn。
用于自組裝的嵌段聚合物還優選具有窄的多分散性指數(pdi),通常是1.01至2.0,更特別是1.01至1.30,再更特別是1.01至1.25。
分層結構
基底是可包含取向控制層(墊層)的分層結構。基底的取向控制層包含共價鍵合形式的連接到基底的底層表面上的墊層的上述無規共聚物。共價鍵合的無規共聚物包含式(a-1)的二價第一重復單元、式(a-2)的二價第二重復單元和式(a-8)的三價第三重復單元:
其中
rz是選自h、甲基、乙基和三氟甲基(*-cf3)的一價基團,
l″是包含1至10個碳的獨立二價連接基,且
k″是二價連接基,且k″的星號鍵共價連接到基底的表面基團上,且
第一重復單元、第二重復單元和第三重復單元無規共價鍵合在無規共聚物的化學結構中。
取向控制層可通過在第一分層結構(第一基底)上布置含有用于墊層的上述無規共聚物、溶劑和任選的選自熱酸發生劑(tags)、光酸發生劑(pags)、催化劑及其組合的一員的溶液并除去溶劑(例如通過熱烘烤和/或暴露在光化光下)形成,由此形成包含頂部取向控制層的第二層結構(第二基底)。取向控制層包含共價鍵合形式的連接到第一分層結構的底層上的無規共聚物。任選地,第二分層結構可以用溶劑沖洗以除去任何未鍵合的無規共聚物。取向控制層對包含聚碳酸酯嵌段的用于自組裝的高chi嵌段共聚物中性潤濕。熱烘烤可以在大約100℃至大約250℃的溫度下進行大約1秒至大約24小時,優選在大約120℃至大約250℃下進行1分鐘至5分鐘。
還公開了用于制備sa層的組合物。組合物包含溶劑、高chi嵌段共聚物和sap。嵌段共聚物和sap溶解在溶劑中。組合物適用于形成包含嵌段共聚物的膜層(sa層)。膜層優選布置在取向控制層(墊層)上。膜層具有與氣氛接觸的頂表面。膜層包含非共價締合的嵌段共聚物和聚合物添加劑。
下列示意圖示出了形成包含用于取向控制的墊層的基底的方法及其在用高chi嵌段共聚物形成垂直取向的層狀結構域圖案中的用途。
圖2.1a至2.1f是顯示包含高chi嵌段共聚物和sap添加劑的sa層的定向自組裝方法的橫截面層圖,其產生垂直取向的層狀結構域,未使用光刻制備的形貌或化學預圖案。應該理解的是,層和特征不按比例繪制。
圖2.1a的分層結構10包含具有基底表面12的基底11。基底11可包含一個或多個層(未示出)。將包含溶解在合適的溶劑中的所公開的用于取向控制的無規共聚物的溶液施加到基底表面12(例如通過旋涂)上,接著除去所有溶劑,產生分層結構20(圖2.1b),也稱作“改性基底”。分層結構20包含用于取向控制的墊層21,其包含通過至少一個共價鍵鍵合到基底11上的無規共聚物。任選地,分層結構20可以用溶劑沖洗以除去任何未鍵合的無規共聚物。
墊層21具有墊層表面22。使用任何合適的技術(例如旋涂)將包含所公開的用于自組裝的含聚碳酸酯嵌段的高chi嵌段共聚物(sa材料)、所公開的sap和溶劑的溶液施加到墊層表面22上。除去溶劑,接著任選后施加烘烤(pab)(例如115℃1分鐘),產生分層結構30(圖2.1c)。分層結構30包含含嵌段共聚物和sap的sa層31。將sa層31布置在墊層表面22上。然后對sa層31施以有效誘發嵌段共聚物自組裝的條件(例如將分層結構30在120℃至250℃的溫度下退火大約1分鐘至大約24小時),由此形成分層結構40(圖2.1d)。分層結構40包含布置在墊層表面22上的自組裝嵌段共聚物的垂直取向層狀結構域圖案41。結構域圖案41包含含有嵌段共聚物的第一嵌段(例如嵌段a,聚苯乙烯)的第一層狀結構域43和含有高chi嵌段共聚物的聚碳酸酯嵌段(例如嵌段b)的第二層狀結構域42。第二層狀結構域42具有比第一層狀結構域43高的sap濃度(未示出)。在這種情況下由嵌段共聚物形成的各結構域的片層與氣氛界面44和墊層表面22接觸。用接觸氣氛界面44的sa層31的頂表面進行sa層的自組裝。
結構域之一可以在其它結構域存在下選擇性除去(例如蝕刻)或改性。作為一個實例,使用合適氣體(o2)的干法蝕刻或濕法/化學蝕刻技術可用于選擇性蝕刻第二層狀結構域42。作為另一實例,第二層狀結構域42(聚碳酸酯嵌段)可通過如下改性第二層狀結構域42而選擇性蝕刻:i)順序滲透合成(sis)以注入金屬氧化物前體或ii)用金屬鹽溶液滲透第二層狀結構域42,接著離子蝕刻。選擇性除去結構域之一也可除去下方的墊層取向控制材料(未示出)。
選擇性除去結構域之一產生包含蝕刻結構域圖案51的分層結構50(圖2.1e)。在這一實例中,蝕刻結構域圖案51包含布置在墊層表面22上的第一層狀結構域43和開口52(已示出)。或者,可以選擇性除去第一層狀結構域43,留下第二層狀結構域42(未示出)。第一層狀結構域43的片層在除去第二層狀結構域42后的尺寸可以不同于它們在選擇性脫除前的尺寸。開口52可具有大約0.5lo的寬度w″(例如對于低chisa材料,w″為大約10納米至大約100納米)。選擇性脫除方法可通過熱烘烤(對于可熱分解材料)、選擇性離子蝕刻法、溶解在選擇性溶劑中或其組合進行。可以通過各種已知方法實現化學改性。例如,結構域可以與硅烷或甲硅烷基氯選擇性反應以將硅含量引入結構域并由此提高其等離子體蝕刻電阻。或者,化學試劑可用于鍵合或化學偶聯到僅位于一種類型的自組裝結構域中的官能團上,以實現例如提高的溶解性質差異,其可有利地用于在其它結構域存在下選擇性除去一種結構域。
最后,可以將蝕刻結構域圖案51轉移到基底11中,由此形成包含轉移圖案61的分層結構60(圖2.1f)。圖案化區域61可以是線、孔、凹坑和/或墊層21和/或基底11的化學改變狀態的圖案,其由區域62表示。圖案化區域61可延伸到一個或多個層,包括墊層21和/或基底11中(已示出)。圖案轉移法可進一步包括除去第一層狀結構域43(未示出)。
圖2.2a至2.2e是顯示光刻法的橫截面層圖,利用與所公開的墊層和包含高chi嵌段共聚物和sap的sa層一起預成型的形貌預圖案。分層結構100(圖2.2a)包含基底110,其包含布置在底層101(例如硅片)的表面103上的墊層102。在墊層表面105上布置形貌預圖案104。墊層102包含通過至少一個共價鍵鍵合到表面103上的一種形式的所公開的無規共聚物。底層101可包含一個或多個子層(未示出)。形貌預圖案104包含形態部106(例如抗蝕形態部)。形態部106具有高度h'的側壁107和寬度w'的頂表面108。形態部106被溝槽109(凹區)分隔,所述溝槽包括含有與氣氛接觸的墊層102的材料的底表面112。預圖案104可通過任何合適的光刻技術形成。形態部106可包含用于引導自組裝的任何合適的材料111。例如,形態部106可包含抗蝕劑材料,其可以是正性和/或負性抗蝕劑材料。
在本發明中,預圖案104的形貌對高chi嵌段共聚物的自組裝層狀結構域的取向控制不是至關重要的。sa層基本或完全位于形態部106的溝槽區109內。形態部106的高度h'通常大于或相當于sa層的厚度。底表面112對sa材料(嵌段共聚物)中性潤濕,而空氣界面對sa材料不是中性的。在這一實例中,側壁107可以對嵌段共聚物中性潤濕或非中性潤濕,只要側壁的表面性質不會不利地影響形成的結構域的自組裝和取向。
使用任何合適的技術(例如旋涂)將包含溶解在溶劑中的sa材料(高chi嵌段共聚物)和sap的涂覆混合物施加到形貌預圖案104上,由此將混合物基本或完全分配在溝槽區109中。形貌預圖案104不溶于或基本不溶于用于制備混合物的溶劑。從施加的涂覆混合物中除去溶劑提供包含sa層121的分層結構120(圖2.2b)。sa層121包含含有sa材料(高chi嵌段共聚物)和sap的區域122。sa層121布置在溝槽區109的底表面112上。
高chi嵌段共聚物的自組裝產生包含垂直取向層狀結構域圖案131的分層結構130(圖2.2c)。自組裝可以是自發的和/或通過熱處理(退火)層121輔助。結構域圖案131包含具有v″的寬度的第一層狀結構域133(例如ps)和具有寬度w″的第二層狀結構域132(例如聚碳酸酯)并布置在溝槽區109的底表面112上。氣氛界面134由箭頭指示。在這一實例中,形態部106的側壁107對第一層狀結構域133具有優先親和性。因此,第一層狀結構域133與側壁107接觸。與側壁107接觸的第一層狀結構域133可具有大約0.5v″的寬度。在一個實施方案中,v″和w″大致等于0.5lo。
結構域之一,例如第二層狀結構域132(例如聚碳酸酯嵌段)可以在第一層狀結構域133(例如ps嵌段)存在下選擇性除去(例如離子蝕刻)或改性以生成形貌或化學對比(contrast)。選擇性除去結構域之一也可除去下方的取向控制材料(未示出),產生包含蝕刻結構域圖案141的分層結構140(圖2.2d)。蝕刻結構域圖案141包含布置在墊層表面112上的第一層狀結構域133、開口142和形態部106。開口142可具有大約0.5lo的寬度w″(例如對于高chi嵌段共聚物,w″可以為大約2納米至大約10納米)。選擇性脫除法可通過熱烘烤(對于可熱分解材料)、選擇性離子蝕刻法、溶解在選擇性溶劑中或其組合進行。可以通過如上所述的各種已知方法實現化學改性。選擇性脫除法可進一步除去形態部106(未示出)。
最后,可以將蝕刻結構域圖案141轉移到墊層102和/或底層101中,由此形成包含圖案化區域151的分層結構150(圖2.2e)。圖案化區域151可以是線、孔、凹坑和/或由改變區域152表示的基底材料的化學改變狀態的圖案。圖案化區域151可延伸到底層101的一個或多個層中。可以例如使用反應性離子蝕刻法實現圖案轉移。可以在形成改變區域152的同時或之后除去形態部106和第一層狀結構域133。轉移后的蝕刻結構域圖案141的高度可以小于轉移前的蝕刻結構域圖案141的高度。
基底,更特別是基底的表面可包含無機或有機材料,如金屬、碳或聚合物。更特別地,基底可包含半導體材料,包括例如si、sige、sigec、sic、ge合金、gaas、inas、inp、氮化硅、氮化鈦、氧化鉿,以及其它iii-v或ii-vi化合物半導體。基底還可包含分層半導體,如si/sige或絕緣體上半導體(soi)。特別地,基底可含有含si半導體材料(即包括si的半導體材料)。半導體材料可以是摻雜、非摻雜的或在其中含有摻雜和非摻雜區。
基底可具有抗反射控制層(arc層)或底部arc層(barc層)以降低薄膜堆疊體的反射率。許多合適的barcs是文獻中已知的,包括單層barcs、雙層barcs、分級barcs和可顯影barcs(dbarcs)。基底還可包含硬掩模、轉移層(例如平坦化層、旋布玻璃層(spin-on-glasslayer)、旋布碳層(spin-oncarbonlayer))和如分層器件所需的其它材料。
輔助聚合物
上文提到的基底、墊層和/或sa層可包括被稱作輔助聚合物的其它聚合物。
輔助聚合物可包含羥基。這些包括羥基封端聚合物(例如羥基封端聚(苯乙烯-co-甲基丙烯酸甲酯),和羥基封端聚(苯乙烯)、羥基封端聚(甲基丙烯酸甲酯)和聚(苯乙烯-b-甲基丙烯酸甲酯)的共混物)、羥基官能化聚合物(例如聚(苯乙烯-co-甲基丙烯酸甲酯-co-甲基丙烯酸2-羥乙酯))。
其它輔助聚合物包括包含反應性基團的材料,如衍生自甲基丙烯酸環氧二環戊二烯酯、甲基丙烯酸縮水甘油酯或肉桂酸乙烯酯的那些。示例性的包含反應性基團的材料包括聚(苯乙烯-co-甲基丙烯酸環氧二環戊二烯酯)、聚(苯乙烯-co-甲基丙烯酸甲酯-co-甲基丙烯酸環氧二環戊二烯酯)、聚(苯乙烯-co-甲基丙烯酸甲酯-co-甲基丙烯酸縮水甘油酯)、聚(苯乙烯-co-甲基丙烯酸甲酯-co-肉桂酸乙烯酯)、聚(苯乙烯-co-甲基丙烯酸甲酯-co-乙烯基苯并環丁烷)和聚(α-甲基苯乙烯-co-甲基丙烯酸甲酯))。反應性聚合物可由于獨自或與附加交聯劑一起的熱或光化學處理而反應。特別地,可以使用催化物類,如強酸性物類來促進反應。強酸性物類可以直接并入涂覆組合物中。或者,可以使用熱酸發生劑或光酸發生劑分子以分別由于熱或光化學處理生成酸性物類。
輔助聚合物的其它非限制性實例包括arc層中所用的材料,其可包括選自聚雙酚、聚砜、聚碳酸酯、聚氫醌、聚鄰苯二甲酸酯、聚苯甲酸酯、聚苯基醚、聚氫醌烷基化物、聚氨基甲酸酯、聚丙二酸酯及其混合物的均聚物和共聚物。通常將這些結構部分官能化以調節聚合物的所需物理性質(光學常數、表面能)。聚合物組分還可含有沿聚合物分布的多個反應性位點以與交聯組分反應。用于arc層的更具體的材料包括聚(4,4′-亞甲基雙酚-co-表氯醇)、聚(4,4′-亞乙基雙酚-co-表氯醇)、聚(4,4′-亞異丙基雙酚-co-表氯醇)、聚(4,4′-亞異丙基雙[2-甲基酚]-co-表氯醇)、聚(4,4′-亞異丙基雙[2,6-二甲基酚]-co-表氯醇)、聚(4,4′-亞環己基雙酚-co-表氯醇)、聚(4,4′-[1-苯基亞乙基]雙酚-co-表氯醇)、聚(4,4′-三氟亞異丙基雙酚-co-表氯醇)、聚(4,4′-六氟亞異丙基雙酚-co-表氯醇)、聚(4,4′-磺酰基雙酚-co-表氯醇)、聚(雙酚af己二酸酯)、聚(雙酚af琥珀酸酯)、聚(二鄰苯二甲酸4,4′-六氟亞異丙基酯-co-表氯醇)、聚(二鄰苯二甲酸4,4′-六氟亞異丙基酯-co-聚(雙酚af))、聚(雙苯甲酸4,4′-六氟亞異丙基酯-co-表氯醇)、聚(3,3′,4,4′-二苯甲酮四甲酸酯-co-表氯醇)、聚(二鄰苯二甲酸4,4′-六氟亞異丙基酯-co-表氯醇-co-2,6-雙[羥甲基]-對甲酚)、聚(3,3′,4,4′-二苯甲酮四甲酸酯-co-表氯醇-co-2,6-雙[羥甲基]-對甲酚)、聚(對苯二甲酸酯-co-表氯醇)、聚(2-硝基對苯二甲酸酯-co-表氯醇)、聚(2-硝基鄰苯二甲酸酯-co-表氯醇)、聚(2-硝基間苯二甲酸酯-co-表氯醇)、聚(氫醌-co-表氯醇)、聚(甲基氫醌-co-表氯醇)、聚(1,2,4-苯三醇-co-表氯醇)、聚(亞甲基-雙[4-氨基苯基]-co-甘油氨基甲酸酯)、聚(亞異丙基-雙[4-氨基苯基]-co-甘油氨基甲酸酯)、聚(亞異丙基-雙[3-羧基-4-氨基苯基]-co-甘油氨基甲酸酯)、聚(亞甲基-雙[4-羥苯基]-co-甘油碳酸酯)、聚(亞異丙基-雙[4-羥苯基]-co-甘油碳酸酯)、聚(亞異丙基-雙[3-羧基-4-羥苯基]-co-甘油碳酸酯)、聚(2-苯基-1,3-丙二醇丙二酸酯)、聚(2-苯基-1,3-丙二醇2-甲基-丙二酸酯)、聚(1,3-丙二醇亞芐基-丙二酸酯)、聚(2-苯基-1,3-丙二醇亞芐基-丙二酸酯)、縮水甘油基封端的聚(雙酚a-co-表氯醇)和來自shinetsu的含硅抗反射涂層a940。另一更具體的輔助聚合物是聚(苯乙烯-co-甲基丙烯酸環氧二環戊二烯酯)無規共聚物p(s-r-edcpma):
其中x和y各自是大于1的整數。另一些輔助聚合物包括聚(苯乙烯-co-甲基丙烯酸甲酯-co-甲基丙烯酸環氧二環戊二烯酯)、聚(苯乙烯-co-甲基丙烯酸甲酯-co-甲基丙烯酸縮水甘油酯)、聚(苯乙烯-co-甲基丙烯酸甲酯-co-甲基丙烯酸2-羥乙酯)、聚(苯乙烯-co-甲基丙烯酸甲酯-co-肉桂酸4-乙烯酯)、聚(苯乙烯-co-甲基丙烯酸甲酯-co-乙烯基苯并環丁烷)、聚(苯乙烯-co-乙烯基苯并環丁烷)、聚(α-甲基苯乙烯-co-甲基丙烯酸甲酯)和聚(甲基戊二酰亞胺)(pmgi)。另一些輔助聚合物包含聚合物刷層,其包括由羥基封端聚(苯乙烯-co-甲基丙烯酸甲酯)、聚(苯乙烯-co-甲基丙烯酸甲酯-co-甲基丙烯酸2-羥乙酯)、羥基封端聚(苯乙烯)、羥基封端聚(甲基丙烯酸甲酯)、聚(苯乙烯-b-甲基丙烯酸甲酯)嵌段共聚物和上述表面親和材料的組合形成的那些。另一些輔助聚合物包括能夠形成自組裝單層的其它嵌段共聚物。
用于制備墊層的涂覆組合物至少包含溶劑和所公開的無規共聚物。
用于制備包含所公開的嵌段共聚物的sa層的涂覆組合物至少包含溶劑、所公開的含聚碳酸酯嵌段的高chi嵌段共聚物和所公開的sap。
上述組合物可另外包含其它材料,包括表面活性劑、聚合物、光酸發生劑和熱酸發生劑。例如,在用于制備墊層的組合物中可包括有機硅酸鹽樹脂。
用于制備墊層和sa層的組合物可通過與微電子制造裝配線中使用的工藝和設備相容的任何合適的方法施加。示例性的非限制性技術包括旋涂、浸涂、刮刀涂覆和噴涂。
用于制備上述聚合物基礎涂覆組合物的示例性非限制性澆鑄溶劑包括甲苯、丙二醇單甲基醚乙酸酯(pgmea)、丙二醇單甲基醚(pgme)、丙酸乙氧基乙酯、苯甲醚、乳酸乙酯、2-庚酮、環己酮、乙酸戊酯、乙酸正丁酯、γ-丁內酯(gbl)、水溶液、丙酮和上述溶劑的組合。
用于墊層的無規共聚物具有3,000至200,000克/摩爾的重均分子量(mw)。類似地,無規共聚物具有1,000至80,000的數均分子量(mn)。無規共聚物還可具有1.01至大約3.0的多分散性(mw/mn)。分子量mw和mn都可通過例如使用通用校準方法(用聚苯乙烯標樣校準)的凝膠滲透色譜法(gpc)測定。
用于定向自組裝的高chi嵌段共聚物(sa材料)具有3,000至200,000克/摩爾的重均分子量(mw)。類似地,高chi嵌段共聚物可具有1,000至80,000的數均分子量(mn)。高chi嵌段共聚物還可具有1.01至3的多分散性(mw/mn)。
用于sa層的表面活性聚合物(sap)具有3,000至200,000克/摩爾的重均分子量(mw)。類似地,sap可具有1,000至80,000的數均分子量(mn)。sap可具有1.01至3的多分散性(mw/mn)。
來自嵌段共聚物薄膜的自組裝結構域的狀態(例如形狀、尺寸和取向)取決于嵌段共聚物構造(二嵌段、三嵌段、星形聚合物、瓶刷嵌段共聚物、雜臂聚合物等)、組成(例如材料、分子量和不同嵌段的體積比)、退火條件(例如溫度、環境和退火時間)、界面性質(例如聚合物-空氣界面和聚合物基底界面)以及限定的幾何(例如薄膜厚度和界限的形貌)。因此,通過調節一個或多個參數,可以根據特定用途的需要調節狀態。
高chi嵌段共聚物的自組裝(即相分離和結構域的排列)可在成膜過程中、在施加后烘烤過程中或在隨后退火過程中發生。合適的退火方法包括熱退火、熱梯度退火、溶劑蒸氣退火或通過其它梯度場退火。更特別地,包含高chi嵌段共聚物的sa層在高于嵌段共聚物的玻璃化轉變溫度(tg)但低于嵌段共聚物的分解或降解溫度(td)的溫度下熱退火。熱退火步驟可以在大約80℃至大約300℃的退火溫度下進行。熱退火可以進行大于0小時至大約100小時,更特別是大約1小時至大約15小時。熱退火的嵌段共聚物自組裝以形成其取向垂直于下方表面平面的有序結構域。一般而言,sa層可具有50至10000埃,更特別是100至5000埃,再更特別100至3000埃的厚度。
嵌段共聚物的兩個有序結構域之間的蝕刻速率差異允許生成附加圖案。通過蝕刻、溶劑或其它手段選擇性除去至少一個自組裝結構域產生納米級凸紋圖案,其包含例如可轉移到下方基底上的孔圖案。蝕刻類型包括在半導體器件制造中使用的任何常見蝕刻,例如干法蝕刻,如等離子體蝕刻或使用選擇性溶劑和/或蒸氣的濕法蝕刻。通常,干法蝕刻法用于蝕刻亞50納米尺寸。在這種圖案顯影/圖案轉移之前,可任選化學改性sa材料的自組裝層以改進圖案轉移所需的性質,如抗蝕刻性或機械性質。
通過選擇性除去結構域之一形成的開口的凸紋圖案可具有大于與高chi嵌段共聚物一起使用的形貌預圖案的空間頻率。
可以將抗蝕材料施加到基底表面、墊層表面、抗蝕形態部的表面和/或嵌段共聚物的結構域圖案上以控制選擇性蝕刻速率。可以通過包括化學氣相沉積(cvd)、等離子體增強cvd、原子層沉積(ald)、連續滲透合成(sis)、金屬鹽的順序滲透、濺射、熱蒸發、電子束蒸發、脈沖激光沉積或與微電子制造中使用的工藝和設備相容的任何合適的沉積方法的方法由氣相沉積抗蝕刻材料。
還公開了包含自組裝高chi嵌段聚合物的薄膜,所述薄膜包含相對于薄膜平面具有垂直取向的層狀結構域。還公開了包含包括墊層的基底和布置在墊層上的自組裝高chi嵌段共聚物薄膜的分層結構,所述薄膜包含相對于薄膜平面具有垂直取向的層狀結構域。在一個實施方案中,分層結構是半導體器件。
上述方法可用于形成包含金屬布線、用于接點或通孔的孔、絕緣區(例如鑲嵌槽或淺槽隔離)和用于適合集成電路器件的設計的電容器結構的溝槽的分層結構。所述方法尤其可用于制造氧化物、氮化物或多晶硅的圖案化層。
上述方法有利地能使自組裝結構具有降低的特征寬度和提高的周期性。結構域特征寬度可以為1納米至大約30納米,5納米至大約18納米,或更特別5納米至大約15納米。
提供下列非限制性實例以進一步例示所公開的聚合物和它們在形成自組裝層中的用途。
實施例
下列實施例中所用的材料列在表1中。
表1.
在此,mn是數均分子量,mw是重均分子量,且mw是一個分子的分子量。
用于自組裝的二嵌段聚合物的制備
二嵌段聚合物(實施例1-3)根據示意26制備。
示意26.
實施例1.用單醇引發劑azps1-oh和rop酸催化劑磷酸二苯酯(dpp)通過碳酸三亞甲酯(tmc)的開環聚合(rop)合成二嵌段聚合物dbp1,n=57,m=75。向配有磁攪拌棒的烘干20毫升圓底燒瓶中,加入azps1-oh(0.70克,0.113毫摩爾,mn=6200,pdi=1.02,n=57,獲自az-electronicmaterials,branchburg,nj)、tmc(1.76克,17.25毫摩爾)和dcm(2.94毫升)。攪拌反應混合物直至azps1-oh大分子引發劑和tmc完全溶解在dcm中,此后加入磷酸二苯酯(dpp,400毫克,1.6毫摩爾,催化劑)。將反應混合物在手套箱中在室溫(r.t.)下攪拌16小時。將反應燒瓶從手套箱中取出并通過將其浸在冰-水浴中冷卻至0℃。通過添加dcm(6毫升)、tea(0.7毫升,02.72毫摩爾)和乙酰氯(0.25毫升,3.52毫摩爾),終止該反應。將該反應在室溫下進一步攪拌2小時。通過在甲醇中沉淀該反應混合物而分離所得聚合物。通過在真空下除去甲醇,將產物收集在燒結玻璃漏斗中,并將所得固體再溶解在thf中以形成20重量%溶液并在甲醇中再沉淀。將固體收集在燒結玻璃漏斗中并在真空下在40℃下干燥2小時以獲得所得化合物。mn(gpc)=17200,mw=17700,pdi=1.029;mn(nmr)=azps(6.2k)-pmtc(7.8k),n=57.4,m=75;tmc%轉化率:大約50%。將所得聚合物溶解在thf中以形成20重量%溶液并在甲醇:乙腈(200毫升,60:40v/v)中沉淀該聚合物。將沉淀的固體和溶劑收集在離心管中并通過在0℃下以4000rpm離心、接著潷析溶劑和在真空爐中在40℃下干燥固體2小時而收集固體,得到dbp1。mn(gpc)=17200,mw=17600,pdi=1.02;mn(nmr)=ps(6.2k)-b-ptmc(7.7k),n=57,m=75。標記ps(6.2k)-b-ptmc(7.7k)是指聚(苯乙烯)嵌段(ps)具有mn=6200且聚(碳酸三亞甲酯)嵌段(ptmc)具有mn=7700。在下列實施例中也使用這一標記法。
實施例2.用dpp催化劑合成dbp2。該二嵌段聚合物用azps2-oh(0.20克,0.02毫摩爾,mn=10000,n=96)、tmc(0.518克,5.078毫摩爾)和dpp(62.5毫克,0.25毫摩爾)使用如實施例1中概述的通用程序制備。mn(gpc)=24900,mw=25800,pdi=1.03;mn(nmr)=ps(10.0k)-b-ptmc(13k),n=96,m=127。
實施例3.用dpp催化劑合成dbp3。該二嵌段聚合物用azps1-oh(0.30克,0.048毫摩爾,mn=6200,n=57)、tmc(0.296克,2.90毫摩爾)和dpp(0.12克,0.483毫摩爾)使用如實施例1中概述的通用程序制備。mn(gpc)=10800,mw=11200,pdi=1.03;mn(nmr)=ps(6.2k)-b-ptmc(2.9k),n=57,m=28。
三嵌段聚合物的合成
實施例4.在4個步驟中進行用dbu合成三嵌段聚合物tbp1。
a)中間聚合物ip-1的制備
向配有攪拌棒的100毫升schlenk燒瓶中加入苯乙烯(14.80克,0.142摩爾)、2-溴異丁酸2-羥乙酯(hebib,0.20克,0.946毫摩爾,atrp引發劑)、cubr(0.126克,0.946毫摩爾)和苯甲醚(15克)。該燒瓶用橡膠隔片密封并通過鼓入氮氣吹掃1小時。此時,加入pmdeta(0.168克,0.946毫摩爾)并將反應燒瓶置于設定在110℃的油浴中280分鐘。通過將該schlenk燒瓶置于冰-水浴中,終止該反應。將該燒瓶向空氣敞開,此時該反應混合物變成深綠色。該混合物通過添加thf(100毫升)稀釋并經過短硅膠柱以除去銅催化劑。所得溶液在真空下濃縮并從thf沉淀在甲醇中兩次。將聚合物ip-1收集在燒結玻璃漏斗中并在真空下在50℃下干燥24小時。mn=5500,mw=6000,pdi=1.07,n=49。
b)中間聚合物ip-2的制備
在500毫升圓底燒瓶中合并i-1(3.0克,0.54毫摩爾)、二甲基甲酰胺(dmf)(100毫升)和疊氮化鈉(0.354克,5.4毫摩爾)并將該反應在80℃下攪拌12小時。然后將該反應混合物冷卻至室溫并在真空下蒸發dmf。將所得固體溶解在thf中并在甲醇中沉淀兩次。將聚合物i-2收集在燒結玻璃漏斗中并在真空下在50℃下干燥24小時。mn=5500,mw=6000,pdi=1.07,n=49。
c)中間聚合物ip-3的制備
向配有磁攪拌棒的100毫升圓底燒瓶中加入ip-2(1.73克,0.314毫摩爾)、炔丙醇(0.14克,2.50毫摩爾)、dmf(12毫升)、pmdeta(90毫克,0.524毫摩爾)和cubr(75毫克,0.524毫摩爾)。將反應混合物在氮氣環境下攪拌24小時。將該燒瓶向空氣敞開,此時該反應混合物變成深綠色。該混合物通過添加thf(100毫升)稀釋并經過短硅膠柱以除去銅催化劑。所得溶液在真空下濃縮并從thf沉淀在甲醇中兩次。將該聚合物收集在燒結玻璃漏斗中并在真空下在50℃下干燥24小時以獲得ip-3mn=6000,mw=6600,pdi=1.07,n=49。
d)三嵌段聚合物tbp1的制備
向配有磁攪拌棒的烘干4毫升小瓶中加入ip-3(0.20克,0.033毫摩爾,mn=6000,pdi=1.07)、碳酸三亞甲酯(tmc,0.47克,4.6毫摩爾)和二氯甲烷(dcm,1.2毫升)。攪拌該反應混合物直至oh-ps-oh大分子引發劑和tmc完全溶解在dcm中,此后加入dbu(25毫克,0.16毫摩爾)。將反應混合物在手套箱中在室溫下攪拌3小時。通過添加dcm(1毫升)、三乙胺(tea,0.2毫升)和乙酰氯(0.1毫升),終止該反應。將該反應在室溫下進一步攪拌2小時。通過在甲醇中沉淀該反應混合物而分離所得聚合物。通過在真空下除去甲醇,將產物收集在燒結玻璃漏斗中,并將所得固體再溶解在thf中以形成20重量%溶液并在甲醇中再沉淀。將固體收集在燒結玻璃漏斗中并在真空下在40℃下干燥2小時以獲得所得化合物。將所得聚合物溶解在thf中以形成20重量%溶液并在甲醇:乙腈(200毫升,60:40v/v)中沉淀該聚合物。將沉淀的固體和溶劑收集在離心管中并通過在0℃下以4000rpm離心、接著潷析溶劑和在真空爐中在40℃下干燥固體2小時而收集固體,得到tbp1。
mn(gpc)=20600,mw=22200,pdi=1.08;
mn(nmr)=ptmc(3.5k)-b-ps(6.0k)-b-ptmc(3.5k)。
以vfptmc表示的ptmc嵌段的體積分數為大約0.48。
實施例5和6.三嵌段聚合物tbp2和tbp3(分別是實施例5和6)根據示意27制備。
示意27.
實施例5,tbp2是使用dpp催化劑的代表。向配有磁攪拌棒的烘干4毫升小瓶中,加入oh-psi1-oh(0.15克,0.013毫摩爾,mn=11,500,n=108,獲自polymersourceincorporated,montreal,canada)、tmc(0.378克,3.70毫摩爾)和dcm(1.85毫升)。攪拌該反應混合物直至ho-psi1-oh大分子引發劑和tmc完全溶解在dcm中,此后加入dpp(65毫克,0.26毫摩爾)。將反應混合物在手套箱中在室溫下攪拌15小時。通過添加dcm(1毫升)、tea(0.2毫升)和乙酰氯(0.1毫升),終止該反應。將該反應在室溫下進一步攪拌2小時。通過在甲醇中沉淀該反應混合物而分離所得聚合物。通過在真空下除去甲醇,將產物收集在燒結玻璃漏斗中,并將所得固體再溶解在thf中以形成20重量%溶液并在甲醇中再沉淀。將固體收集在燒結玻璃漏斗中并在真空下在40℃下干燥2小時以獲得所得化合物。將所得聚合物溶解在thf中以形成20重量%溶液并在甲醇:乙腈(200毫升,60:40v/v)中沉淀該聚合物。將沉淀的固體和溶劑收集在離心管中并通過在0℃下以4000rpm離心、接著潷析溶劑和在真空爐中在40℃下干燥固體2小時而收集固體。
mn(gpc)=27900,mw=30300,pdi=1.09;
mn(nmr)=ptmc(7.05k)-b-ps(11.5k)-b-ptmc(7.05k),n=108,m=138。
vfptmc~0.49。
實施例6.用dpp催化劑合成tbp3。該聚合物用ho-psi2-oh(0.15克,0.0095毫摩爾,mn15700,n=148)、tmc(0.155克,1.528毫摩爾)和dpp(24毫克,0.095毫摩爾)使用如實施例5中概述的通用程序制備。
mn(gpc)=24200,mw=26400,pdi=1.09;
mn(nmr)=ptmc(3.5k)-b-ps(14.7k)-b-ptmc(3.5k),n=148,m=68。
vfptmc~0.26。
表2總結了實施例1-6的嵌段共聚物的制備和性質。
表2.
基于苯乙烯、tmc和gma的墊層無規共聚物
這些無規共聚物(實施例7-18)在名稱中具有前綴"g1"。
實施例7.無規共聚物p-1的合成,sty:hema:gma摩爾比x:y:z=88:6:6,sty:hema:gmadp比率x':y':z'=50.5:3.5:3.5
在上述標記法中,方括號內的重復單元的豎直堆疊表示該聚合物鏈中的重復單元的無規分布。端基e'連接到與左方括號搭接的星號鍵之一上。端基e″連接到與右方括號搭接的星號鍵之一上。應該理解的是,對于給定重復單元,與左方括號搭接的星號鍵可連接到在與右方括號搭接的右星號鍵所示的位置處的不同重復單元上,或連接到端基e'上。同樣地,對于給定重復單元,與右方括號搭接的星號鍵可連接到在與左方括號搭接的左星號鍵所示的位置除處的不同重復單元上,或連接到端基e″上。除非另行指明,下標x'、y'和z'代表相應括號內的重復單元在該聚合物中的平均數。對于p-1,未示出端基e'和e″。
在配有磁攪拌棒和頂置冷凝器的250毫升圓底燒瓶(rbf)中合并苯乙烯(sty,14.4克,138.0毫摩爾)、甲基丙烯酸羥乙酯(hema,1.0克,7.68毫摩爾)、甲基丙烯酸縮水甘油酯(gma,1.09克,7.66毫摩爾)、thf(50克)和偶氮二異丁腈(aibn,0.757克,4.61毫摩爾,乙烯基單體的總摩爾數的3摩爾%)。將反應混合物在70℃下攪拌18小時并通過將該反應冷卻至室溫停止。所得聚合物通過在meoh中沉淀兩次分離,并在真空下在50℃下干燥24小時。mn=6200,mw=8700,pdi=1.40。通過13c反門控nmr計算產物sty:hema:gma摩爾比x:y:z為x:y:z=88:6:6。基于mn和產物摩爾比,計算p-1的各重復單元sty:hema:gma的聚合度(dp),分別為x':y':z'=50.5:3.5:3.5。
實施例8.由大分子引發劑p-1(實施例7,sty:hema:gma摩爾比x:y:z=88:6:6,dp比x':y':z'=50.5:3.5:3.5)合成tmc-官能無規接枝共聚物g1-1以用于取向控制
量y'a'(即y'乘以a')代表無規接枝共聚物g1-1中衍生自tmc的重復單元的總平均數。在這些計算中,在測定各重復單元的dp時,未對端基e'和e″的質量調節mn。
將p-1(0.2克,實施例6)、碳酸三亞甲酯(tmc,0.060克,0.588毫摩爾)和二氯甲烷(dcm,0.2克)添加到配有磁攪拌棒的烘干4毫升玻璃小瓶中。攪拌該反應混合物直至大分子引發劑和tmc完全溶解在dcm中,此后加入1,8-二氮雜雙環[5.4.0]十一碳-7-烯(dbu,~10毫克)。將反應混合物在手套箱中在室溫(r.t.)下攪拌1小時。通過將反應小瓶從手套箱中取出并通過添加dcm(0.5毫升)、三乙胺(tea,0.27克,2.72毫摩爾)和乙酰氯(~60毫克,0.764毫摩爾),終止該反應。將該反應在室溫下進一步攪拌2小時。通過在甲醇中沉淀該反應混合物而分離所得聚合物。通過在真空下除去甲醇,將產物收集在燒結玻璃漏斗中,并將所得固體再溶解在thf中以形成20重量%溶液并在甲醇中再沉淀兩次。將固體收集在燒結玻璃漏斗中并在真空下在40℃下干燥2小時以獲得無規接枝共聚物g1-1,其中通過1hnmr測定sty:tmc摩爾比x':y'a'=73:27。如下計算a'的平均值:
x'/y'=50.5/3.5(p-1大分子引發劑的dp比),
x'/(y'a')=73/27(通過1hnmr得出的g1-1的sty:tmc摩爾比),
重排,x'/y'=73a'/27
代入,50.5/3.5=73a'/27,因此
解得,a'=5.33。
因此,g1-1具有碳酸酯重復單元平均數a'=5.33的側鏈聚碳酸酯,基于g1-1的mn。
實施例9-18.制備用于取向控制的墊層無規接枝共聚物g1-2至g1-11。使用實施例8的通用程序和大分子引發劑p-1(實施例7)在各種sty:tmc摩爾比下制備這些聚合物。g1-2至g1-11與g1-1的區別在于a',側鏈中的tmc重復單元平均數。結果總結在表3中。
表3.
1a'基于無規接枝共聚物的mn,未校正端基
基于苯乙烯、acema和gma的墊層的合成
這些無規共聚物(實施例19-25)在名稱中具有前綴"ghs"并具有式結構
其中重復單元平均數x'、y'和z'大于0。未示出端基e'和e″。
實施例19.聚(苯乙烯-r-甲基丙烯酸乙酰氧基乙酯-r-甲基丙烯酸縮水甘油酯)墊層ghs-1的合成,x:y:z=77:19:4(摩爾比),dp比x':y':z'=48.5:12.1:2.6.
在配有磁攪拌棒和頂置冷凝器的250毫升圓底燒瓶(rbf)中合并苯乙烯(sty,4.0克,38.4毫摩爾,mw104.2)、甲基丙烯酸乙酰氧基乙酯(acema,1.21克,7.02毫摩爾,mw172.2)、甲基丙烯酸縮水甘油酯(gma,0.20克,1.40毫摩爾,mw142.2)、thf(22克)和偶氮二異丁腈(aibn,0.31克,1.87毫摩爾,乙烯基單體的總摩爾數的4摩爾%)。將反應混合物在70℃下攪拌18小時并通過將該反應冷卻至室溫停止。所得聚合物通過在meoh中沉淀兩次分離,并在真空下在50℃下干燥24小時,得到ghs-1。mn=7500,mw=10,100,pdi=1.34。通過13c反門控nmr計算sty:acema:gma的產物摩爾比為x:y:z=77:19:4(摩爾比)。基于mn并且未對端基e'和e″調節,sty:acema:gma的聚合度比率為x':y':z'=48.5:12.1:2.6或四舍五入至零小數位為48:12:3。
實施例20-25.制備用于墊層取向控制的無規接枝共聚物ghs-2至ghs-7。使用實施例19的通用程序在如表4中總結的各種sty:acema:gma摩爾比下制備這些聚合物。
表4.
表面活性聚合物添加劑(sap)的合成
實施例26.合成sap1,hfa-sty均聚物
未示出sap1的端基e'和e″。在配有磁攪拌棒和頂置冷凝器的100毫升圓底燒瓶(rbf)中合并六氟醇苯乙烯(hfa-sty,5.28克,19.54毫摩爾,mw270.2)、thf(16克)和偶氮二異丁腈(aibn,0.182克,1.10毫摩爾,乙烯基單體的總摩爾數的4摩爾%)。將反應混合物在70℃下攪拌18小時并通過將該反應冷卻至室溫停止。所得聚合物通過在己烷中沉淀兩次分離,并在真空下在50℃下干燥24小時。mn=5800,mw=12300,pdi=2.13。基于mn并且未對端基e'和e″調節,聚合度x'=21。
實施例27.合成sap2,bishfachma均聚物
未示出sap2的端基e'和e″。根據實施例26的通用程序使用bishfachma(1.0克,2.19毫摩爾,mw456.1)、thf(4.0克)和偶氮二異丁腈(aibn,0.014克,0.0876毫摩爾,乙烯基單體的總摩爾數的4摩爾%)制備sap2。mn=8200,mw=11100,pdi=1.34。基于mn而不調節端基e'和e″,聚合度x'=18。
實施例28.合成sap3,iprhfama均聚物
未示出sap3的端基e'和e″。根據實施例26的通用程序使用iprhfama(5.0克,17.0毫摩爾,mw456.1)、thf(15.0克)和偶氮二異丁腈(aibn,0.11克,0.68毫摩爾,乙烯基單體的總摩爾數的4摩爾%)制備sap3。mn=8400,mw=14500,pdi=1.73。基于mn而未對端基e'和e″調節,聚合度x'=18。
實施例29.合成sap5-20,苯乙烯和hfa-sty的無規共聚物
在配有磁攪拌棒和頂置冷凝器的100毫升圓底燒瓶(rbf)中合并苯乙烯(sty,0.29克,2.78毫摩爾,mw104.2)、六氟醇苯乙烯(hfa-sty,3.0克,11.1毫摩爾,mw270.2)、thf(10克)和偶氮二異丁腈(aibn,0.091克,0.55毫摩爾,乙烯基單體的總摩爾數的4摩爾%)。將反應混合物在70℃下攪拌18小時并通過將該反應冷卻至室溫停止。所得聚合物通過在己烷中沉淀兩次分離,并在真空下在50℃下干燥24小時。mn=13100,mw=22900,pdi=1.73。通過13c反門控nmr計算產物sty:hfa-styx:y摩爾比為x:y=22:78(摩爾比)。基于mn而未對端基e'和e″調節,dp比x':y'=12.3:43.7或12:44。
實施例30.合成sap6-20,五氟苯乙烯(pfs)和hfa-sty的無規共聚物
在配有磁攪拌棒和頂置冷凝器的100毫升圓底燒瓶(rbf)中合并五氟苯乙烯(pfs,0.54克,2.78毫摩爾,mw194)、六氟醇苯乙烯(hfa-sty,3.0克,11.1毫摩爾,mw270.2)、thf(10克)和偶氮二異丁腈(aibn,0.091克,0.55毫摩爾,乙烯基單體的總摩爾數的4摩爾%)。將反應混合物在70℃下攪拌18小時并通過將該反應冷卻至室溫停止。所得聚合物通過在己烷中沉淀兩次分離,并在真空下在50℃下干燥24小時。mn=14500,mw=26700,pdi=1.84。通過13c反門控nmr計算產物pfs:hfa-styx:y摩爾比為x:y=17:83(摩爾比)。基于mn和排除端基e'和e″,dp比x':y'=9.6:46.5。
實施例31-33.分別合成sap6-40至sap6-80。使用實施例30的通用程序在pfs和hfa-sty的不同摩爾比下制備pfs和hfa-sty的這些無規共聚物(見表5)。
實施例34.合成pf-oibr,氟化原子轉移自由基聚合(atrp)引發劑
在配有磁攪拌棒的250毫升圓底燒瓶中合并1h,1h-全氟壬-1-醇(pf-oh,3.0克,6.66毫摩爾)、三乙胺(tea,1.01g,1.0毫摩爾)和thf(50毫升)。在室溫下經30分鐘向這種混合物中逐滴加入溴異丁酰溴(bribr,1.83克,8.0毫摩爾)在thf(20毫升)中的溶液。將反應混合物在氮氣下攪拌24小時,此后通過使其暴露在空氣中而終止反應。該溶液經濾紙過濾以除去三乙胺氫溴酸鹽,并在真空下除去thf。所得粗制化合物使用硅膠柱色譜法用己烷:乙酸乙酯(80:20)混合物提純以獲得純全氟標記atrp引發劑pf-oibr。
實施例35.合成sap7-c,具有多氟化端基的hfa-sty的均聚物,通過atrp聚合制備
向配有攪拌棒的50毫升schlenk燒瓶中加入hfa-sty(3.0克,11.1毫摩爾,mw270.2)、atrp引發劑pf-oibr(0.443克,0.74毫摩爾,mw598,實施例32)、cubr(0.106克,0.74毫摩爾)和苯甲醚(3.0克)。該燒瓶用橡膠隔片密封并通過鼓入氮氣吹掃1小時。此時,加入pmdeta(0.128克,0.74毫摩爾)并將反應燒瓶置于設定在80℃的油浴中100分鐘。通過將該schlenk燒瓶置于冰-水浴中,終止該反應。將該燒瓶向空氣敞開,此時該反應混合物變成深綠色。該混合物通過添加thf(100毫升)稀釋并經過短硅膠柱以除去銅催化劑。所得溶液在真空下濃縮并從thf沉淀在冷己烷中兩次。將該聚合物收集在燒結玻璃漏斗中并在真空下在50℃下干燥24小時。mn=3200,mw=3600,pdi=1.08。基于mn,x'=9.6。
表5總結了表面活性聚合物實施例26-33的制備,通過使用aibn引發劑的自由基聚合制成。
表5.
表6總結了表面活性聚合物實施例26-33和35的nmr和gpc分析。
表6.
墊層和復合層膜制備
實施例36-42.用無規接枝共聚物g1-2、g1-3,g1-7,g1-8和g1-10(見上表3)薄膜制備墊層(uls)
使用下列通用程序在硅片上制備薄膜墊層。通過將無規接枝共聚物(95重量份)和對硝基芐基磺酸三氟甲磺酸鹽(p-nbt,5重量份)溶解在丙二醇單甲基醚乙酸酯(pgmea,10,000重量份)中而制備溶液,以形成基于總干固體計1.0重量%溶液。p-nbt是熱酸發生劑并為促進在烘烤(退火)時無規接枝共聚物的薄膜在硅片基底上的接枝和部分交聯而加入。使溶液經過0.2mm聚四氟乙烯(ptfe)過濾器,然后以2000rpm旋轉速率在硅片上旋涂該溶液。在形成薄膜后,該涂覆硅片在200℃下烘烤3分鐘并冷卻至室溫。初始烘烤薄膜(墊層)具有用nanospecreflectometer測得的20納米厚度。然后通過在涂覆硅片上澆注pgmea而對墊層進行溶劑沖洗,使溶劑攪動(puddle)30秒,并將處理過的硅片在2000rpm下旋轉干燥30秒。該沖洗旨在除去未交聯或接枝到硅片表面上的所有過量無規接枝共聚物。在溶劑沖洗后墊層的最終薄膜厚度為10納米。表7總結了由名稱中具有"g1"前綴的無規共聚物制成的墊層膜ul-1至ul-5和ul-13至ul-14。
表7.
1x'、y'和a'基于無規接枝共聚物的mn
實施例43-49.使用無規共聚物ghs-1至ghs-7(見上表4)薄膜制備墊層(uls)
使用下列通用程序在硅片上制備薄膜墊層。通過將無規接枝共聚物(95重量份)和對硝基芐基磺酸三氟甲磺酸鹽(p-nbt,5重量份)溶解在丙二醇單甲基醚乙酸酯(pgmea,10,000重量份)中而制備溶液,以形成基于總干固體計1.0重量%溶液。p-nbt是熱酸發生劑并為促進在烘烤(退火)時無規接枝共聚物的薄膜在硅片基底上的接枝和部分交聯而加入。使溶液經過0.2mm聚四氟乙烯(ptfe)過濾器,然后以2000rpm旋轉速率在硅片上旋涂該溶液。在形成薄膜后,該涂覆硅片在190℃下烘烤3分鐘并冷卻至室溫。初始烘烤薄膜(墊層)具有用nanospecreflectometer測得的20納米厚度。然后通過在涂覆硅片上澆注pgmea而對墊層進行溶劑沖洗,使溶劑攪動(puddle)30秒,并將處理過的硅片在2000rpm下旋轉干燥30秒。該沖洗旨在除去未交聯或接枝到硅片表面上的所有過量無規接枝共聚物。在溶劑沖洗后墊層的最終薄膜厚度為10納米。表8總結了分別用ghs-1至ghs-7制成的墊層膜ul-6至ul-12。
表8.
嵌段共聚物和sap添加劑的配制
實施例50-91.含有實施例1和3-6的嵌段共聚物和實施例26-33和35的表面活性聚合物(sap)的用于自組裝的涂覆制劑的制備
下列通用程序是代表性的。制備單獨的嵌段共聚物溶液用于一次涂覆。將用于自組裝的嵌段共聚物(0.01克)溶解在pgmea(0.823克)以形成該嵌段共聚物的1.2重量%溶液。將表面活性聚合物(0.1克,sap實施例26-33和實施例35)溶解在pgmea(8.23克)中以形成基于該溶液的總重量計1.2重量%sap聚合物儲液。該sap溶液經0.2微米ptfe過濾器過濾。然后將一部分sap儲液添加到整個嵌段共聚物溶液中以形成含有所需sap濃度的涂覆組合物。此溶液在室溫下攪拌以形成均勻混合物,此后其經0.2微米ptfe過濾器過濾。表9概括的制成的含有sap添加劑的嵌段共聚物涂覆制劑。
表9.
配制的嵌段共聚物組合物在ul-1至ul-14上的薄膜自組裝
實施例92-177.使用下列通用程序在ul-1至ul-14基底上制備配制的嵌段共聚物的薄膜。以2000rpm的旋轉速率在實施例36-49的所選墊層涂覆基底上旋涂實施例50-91中制成的所選涂覆制劑。然后將涂覆晶片在140℃或170℃的溫度下烘烤(退火)5分鐘并立即冷卻至室溫。通過原子力顯微術(afm)表征配制的嵌段共聚物薄膜,其中使用具有以輕敲模式運行的1n/m彈簧常數氮化硅懸臂的digitalinstruments3100afm。將掃描尺寸和速度分別設定為2微米x2微米面積和1hz。表10總結了實施例92-177中制備的薄膜的涂覆和退火條件。
表10.
表11總結了在各種墊層上制成的自組裝膜的性質和通過自組裝獲得的狀態。“i/h”是指島/孔(不合意)。“部分⊥片層”是指大約20%至小于70%的薄膜區域含有垂直片層(不合意)。“大體上⊥片層”是指70%至小于95%的薄膜區域含有垂直片層(合意)。“ll圓柱體”是指平行圓柱體(不合意)。“⊥片層”是指95%至100%的薄膜區域含有垂直片層(最合意)。“平坦”是指在該膜中沒有可識別的結構。
表11.
圖3-84分別是實施例92-177的自組裝嵌段共聚物膜的原子力顯微術(afm)圖像。sap1、sap6-40和末端官能化的sap7-c是優選的sap組成。表11的結果表明使用基于用于制備sa層的制劑的總干固體計2重量%至10重量%的sap濃度形成垂直片層。sap6-40和sap7-c在以用于制備sa層的制劑的總干固體的2重量%至3重量%的量使用時有效產生垂直片層。在實施例175中0.5重量%的sap7-c濃度足以獲得垂直圓柱體。
制圖外延法定向自組裝(dsa)
實施例178-181.在這些實施例中,使用負性光致抗蝕劑在中性墊層(ul-4)上形成形貌預圖案,接著在該預圖案上施涂配制的嵌段共聚物薄膜。該嵌段共聚物基本局限在抗蝕預圖案的溝槽中。然后將該涂覆結構退火,以使該預圖案引導嵌段共聚物的自組裝。
下述程序是代表性的。通過將無規接枝共聚物g1-8(0.095克,95重量份)和對硝基芐基磺酸三氟甲磺酸鹽(p-nbt,0.005克,5重量份)溶解在丙二醇單甲基醚乙酸酯(pgmea,9.90克,10,000重量份)中而制備墊層溶液,以形成基于該溶液的總重量計1.0重量%溶液。添加熱酸發生劑p-nbt以促進無規接枝共聚物膜在硅片基底堆疊體上的接枝和部分交聯。該硅片基底堆疊體包含硅片底層,其被~30納米厚非晶碳層和10納米厚氮化硅(sinx)層涂覆。使墊層溶液經過0.2微米聚四氟乙烯(ptfe)過濾器,然后以2000rpm旋轉速率在硅片堆疊體上旋涂該溶液。在形成膜后,將該涂覆硅片在200℃下烘烤3分鐘并冷卻至室溫。初始烘烤薄膜(墊層)具有用nanospecreflectometer測得的20納米厚度。然后通過在涂覆硅片上澆注pgmea而對墊層進行溶劑沖洗,使溶劑攪動(puddle)30秒,并將處理過的硅片在2000rpm下旋轉干燥30秒。該沖洗旨在除去未交聯或接枝到硅片表面上的所有過量無規接枝共聚物。在溶劑沖洗后墊層的最終薄膜厚度為10納米。
接著,在這一墊層涂覆基底上布置商業193nm負性光致抗蝕劑(jsrarf7210jn-8)的60納米厚的層,接著在80℃下進行施涂后烘烤60秒。該光致抗蝕劑層然后使用193nm浸漬干涉工具(ibmnemo)以4.67mj的固定劑量曝光,在95℃下烘烤60秒,并用2-庚酮顯影劑顯影60秒。所得200納米印刷密度(pitch)圖案化的光致抗蝕劑層然后在200℃下烤硬3分鐘,然后涂覆嵌段共聚物制劑。
如下制備嵌段共聚物制劑。將二嵌段共聚物dbp1(0.01克)溶解在pgmea(1.24克,10,000重量份)中以形成基于該溶液的總重量計0.8重量%的嵌段共聚物儲液。使該溶液經過0.2微米聚四氟乙烯(ptfe)過濾器。通過將所選sap添加劑(0.1克,見表12)以該溶液總重量的0.8重量%sap溶解在pgmea(12.4克)中,制備單獨的儲液。使該sap溶液經過0.2微米ptfe過濾器。將所需量的sap儲液(表12)添加到嵌段共聚物溶液中并充分攪拌該混合物以形成均勻溶液。將含有嵌段共聚物和sap的所得溶液旋涂在上述圖案化光致抗蝕劑基底上。在旋涂后,該涂覆硅片在表9中規定的溫度下烘烤表9中規定的時間,并立即冷卻至室溫。用自頂向下和橫截面sem分析該導向預圖案溝槽內的嵌段共聚物的自組裝結構域。對于自頂向下sem,樣品在頂表面上使用rie用四氟乙烯(cf4/h2)氣體蝕刻5秒,對于橫截面sem,在該膜的橫截面上施以垂直蝕刻8秒。在sem成像前對樣品施以使用20ma電流的20秒au濺射。
表12總結了嵌段共聚物制劑和在預圖案基底上的涂覆條件。
表12.
sap6-40和末端官能化sap7-c是用于定向自組裝的優選sap組成。
實施例178(圖85a和85b,sems)和實施例179在預圖案的溝槽內形成垂直取向片層。但是,片層相對于預圖案表現出無規橫向排列。實施例99和103分別對應于實施例178和179,無預圖案。
實施例180(圖86a和86b,sems)和實施例181(圖86c)在預圖案的溝槽內形成垂直取向片層。片層也平行于導向預圖案橫向排列。上述實施例139對應于實施例180,但無預圖案并具有略低sap重量%。沒有制備對應于實施例181的無預圖案的實施例。
無sap添加劑的嵌段共聚物制劑
實施例182-187(對比).使用實施例1-6的嵌段共聚物制備嵌段共聚物涂覆制劑但未添加sap添加劑。下列通用程序是代表性的。如上所述制備嵌段共聚物在pgmea中的1.2重量%溶液,所得溶液經0.2微米ptfe過濾器過濾。表13總結了不用sap添加劑制備的嵌段共聚物制劑。
表13.
無sap添加劑的嵌段共聚物制劑的薄膜自組裝
實施例188-204(對比).使用下列通用程序在各種墊層涂覆基底上制備不含sap添加劑的嵌段共聚物制劑實施例181-187的薄膜。以所需旋轉速率(表14)在墊層涂覆基底上旋涂涂覆制劑溶液。在形成薄膜后,涂覆硅片在所需時間和溫度下烘烤并立即冷卻至室溫。通過使用具有以輕敲模式運行的1n/m彈簧常數氮化硅懸臂的digitalinstruments3100afm的原子力顯微術(afm)表征嵌段共聚物薄膜。將掃描尺寸和速度分別設定為2微米x2微米面積和1hz。表14總結了所用涂覆和退火條件和在自組裝后的嵌段共聚物薄膜的所得狀態。
表14.
表14的所有薄膜表現出島/孔或平行圓柱體狀態(不合意)。圖87-89分別是實施例188-190的afm高度圖像。圖90-91分別是實施例194-195的afm高度圖像。圖92-96分別是實施例200-204的afm高度圖像。
油酸作為表面活性材料
實施例205-211(對比).在具有墊層ul-3的基底上用油酸或十氟辛二酸(dfs)作為添加劑配制的嵌段共聚物組合物的薄膜制備和表征
使用下列通用程序在ul-3基底上制備用油酸或十氟辛二酸(dfs)配制的二嵌段共聚物dbp1(實施例1)的薄膜。通過將該嵌段共聚物(0.01克)溶解在pgmea(0.823克)中而制備dbp1溶液,以形成基于總干固體計1.2重量%溶液。使該溶液經過0.2微米聚四氟乙烯(ptfe)過濾器。將所需量的油酸或十氟辛二酸(dfs)添加到上文制成的嵌段共聚物溶液中并充分攪拌該混合物以形成均勻溶液,然后以所需旋轉速率在墊層涂覆基底上旋涂該配制的嵌段共聚物溶液。在形成薄膜后,該涂覆硅片在所需時間和溫度下烘烤并立即冷卻至室溫。通過使用具有以輕敲模式運行的1n/m彈簧常數氮化硅懸臂的digitalinstruments3100afm的原子力顯微術(afm)表征配制的嵌段共聚物薄膜。將掃描尺寸和速度分別設定為2微米x2微米面積和1hz。表15總結了在ul-3上的配制嵌段共聚物薄膜自組裝和所得狀態。表15的所有樣品表現出島/孔(i/h)狀態。圖97-103分別是實施例205-211的afm高度圖像。
表15.
這些結果表明只有氫鍵合oh基團(在這一實施例中,羧酸基)的表面活性材料與包含氟和氫鍵合oh基團的表面活性材料相比不能有效形成垂直取向片層。
實施例212(對比).合成用作墊層(ul-15)的sap6-40a
未示出端基e'和e″。在配有磁攪拌棒和頂置冷凝器的100毫升圓底燒瓶(rbf)中合并五氟苯乙烯(pfs,0.91克,4.69毫摩爾)、苯乙烯六氟醇(hfa-sty1.80克,6.68毫摩爾)、甲基丙烯酸縮水甘油酯(gma,0.05克,0.351毫摩爾)、thf(8克)和偶氮二異丁腈(aibn,0.077克,0.461毫摩爾,乙烯基單體的總摩爾數的4摩爾%)。將反應混合物在70℃下攪拌18小時并通過將該反應冷卻至室溫停止。所得聚合物通過在meoh中沉淀兩次分離,并在真空下在50℃下干燥24小時。mn=16300,mw=27000,pdi=1.65。
實施例213(對比).使用sap6-40a(實施例212)制備墊層ul-15
通過將實施例212的無規接枝共聚物sap6-40a(95重量份,0.095克)和p-nbt(5重量份,0.005克)溶解在pgmea(9.90g)中而制備溶液,以形成基于總干固體計1.0重量%溶液。使該溶液經過0.2微米聚四氟乙烯(ptfe)過濾器,然后以2000rpm(轉/分鐘)旋轉速率在硅片上旋涂該溶液。在形成薄膜后,該涂覆硅片在190℃下烘烤3分鐘并冷卻至室溫。然后通過在涂覆硅片上澆注pgmea而對墊層進行溶劑沖洗,使溶劑攪動(puddle)30秒,并將處理過的硅片在2000rpm下旋轉干燥30秒。該沖洗旨在除去未交聯或接枝到硅片表面上的所有過量無規接枝共聚物。
實施例214-215(對比).在墊層ul-15上的無sap添加劑的二嵌段共聚物dbp1(實施例1)薄膜制備和表征
將實施例1的二嵌段共聚物dbp1(0.01克)溶解在pgmea(0.823克)中以形成基于總干固體計1.2重量%溶液。使該溶液經過0.2微米聚四氟乙烯(ptfe)過濾器。在兩種不同的旋轉速率(2000rpm和3000rpm)下在實施例213的墊層(ul-15)涂覆基底上旋涂該溶液以形成兩個樣品。在形成薄膜后,涂覆硅片在所需時間和溫度下烘烤并立即冷卻至室溫。通過使用具有以輕敲模式運行的1n/m彈簧常數氮化硅懸臂的digitalinstruments3100afm的原子力顯微術(afm)表征嵌段共聚物薄膜。將掃描尺寸和速度分別設定為2微米x2微米面積和1hz。表16總結了在ul-15上的二嵌段共聚物薄膜自組裝和所得狀態。圖104-105分別是實施例214-215的afm高度圖像。各自組裝層表現出島/孔狀態,表明用sap添加劑(sap6-40)制成的墊層對dbp1非中性。
表16.
實施例216.實施例102的薄膜的gisaxs表征
使用gisaxs(掠入射小角度x射線散射)證實在大面積(50微米x毫米)上的實施例102的薄膜的取向。在測量過程中樣品垂直于檢測器放置。在傅里葉空間(圖106c,曲線圖)中繪制yoneda帶上方的背景校正積分散射強度。圖106a和圖106b分別顯示樣品的相應sem圖像,自頂向下和橫截面。在1:2:3的逆空間比率下可以觀察到尖銳一階散射峰和較弱的二階和三階峰,這是垂直取向片層納米結構特征性的。這表明自組裝沒有在空氣界面形成添加劑層,且層狀結構域與空氣界面接觸。在
實施例217.ptmc均聚物的合成
向配有磁攪拌棒的烘干4毫升玻璃小瓶中加入購得的tmc(1.0克,9.8毫摩爾)、芐基醇(bnoh,10毫克,0.092毫摩爾)和dcm(2.4毫升)。攪拌該反應混合物直至tmc完全溶解在dcm中,此后加入dbu(22毫克,0.147毫摩爾)。將反應混合物在手套箱中在室溫下攪拌4小時。通過將反應小瓶從手套箱中取出和通過添加乙酰氯(30毫克,0.384毫摩爾)并將反應混合物在室溫下進一步攪拌45分鐘,終止該反應。使該聚合物在冷甲醇(0℃)中沉淀。潷析甲醇并將該聚合物在真空爐中在室溫下干燥。該干燥聚合物通過溶解在thf中和在冷甲醇(0℃)中再沉淀而進一步提純。潷析甲醇并將該聚合物在真空爐中在室溫下干燥以獲得最終產物。mn=15.0k,mw=16.1k,pdi=1.08,n=147。
實施例218-226用于afm/粘合試驗樣品的ps均聚物、ptmc均聚物和bcp+sap的薄膜制備
a)使用無規聚(苯乙烯-r-甲基丙烯酸甲酯-r-甲基丙烯酸縮水甘油酯)共聚物(ps-pmma-gma,58/40/2摩爾比)制備墊層ul-16。將無規共聚物ps-pmma-gma(0.095克)和p-nbt(0.005克)溶解在pgmea(9.90克)中以形成基于總干固體計1.0重量%溶液。p-nbt是熱酸發生劑并為促進該無規接枝共聚物在硅片上的接枝和部分交聯而加入。使該溶液經過0.2微米聚四氟乙烯(ptfe)過濾器,然后以3000rpm旋轉速率旋涂該溶液。在形成薄膜后,該涂覆硅片在215℃下烘烤2分鐘并冷卻至室溫。然后通過在涂覆硅片上澆注pgmea而對墊層進行溶劑沖洗,使溶劑攪動(puddle)30秒,并將處理過的硅片在2000rpm下旋轉干燥30秒。該沖洗旨在除去未交聯或接枝到硅片表面上的所有過量無規接枝共聚物。在溶劑沖洗后墊層的最終薄膜厚度為7納米。
b)聚苯乙烯均聚物(ps)、聚(碳酸三亞甲酯)均聚物(ptmc)和bcp+sap的薄膜制備。在墊層ul-16上制備聚(苯乙烯)均聚物(mw=5000da,polymersource)、二嵌段共聚物dbp1(實施例1)和sap6-40(實施例31)的薄膜。在硅片上制備聚(碳酸三亞甲酯)(ptmc,實施例217,mn15000)的薄膜。加工條件總結在表17中。
表17.
制成的薄膜然后通過afm表征。用具有以輕敲模式運行的3.8211n/m彈簧常數氮化硅懸臂的digitalinstruments3100afm進行afm表征。表征結果總結在表18中。
表18.
實施例221、222和223的薄膜(dbp1+sap6-40)的粘合值類似于實施例218和219(ps均聚物)。ptmc具有與尖端(tip)的高粘合,而ps具有相對較低的與尖端(tip)的粘合。sap對尖端(tip)具有最低粘合響應。無sap以及具有各種量的sap添加劑的ps結構域產生類似的粘合響應,表明sap添加劑沒有富集在自組裝dbp1的ps結構域的頂表面,而是ptmc結構域的頂表面。與實施例225的純ptmc均聚物的值相比,實施例226(ptmc+sap)和實施例221、222和223(嵌段共聚物dbp1+sap)的粘合值的顯著降低進一步證實sap選擇性富集在ptmc結構域的頂表面的假說。
實施例227-232.bcp+sap薄膜的sims表征
使用下列通用程序在ul-2基底上制備配制的嵌段共聚物的薄膜。將嵌段共聚物dbp1(0.01克,實施例1)溶解在pgmea(0.823克)中以形成基于總干固體計1.2重量%溶液。使該溶液經過0.2微米聚四氟乙烯(ptfe)過濾器。在pgmea中制備實施例26、31和33的具有1.2重量%sap添加劑的單獨儲液并經過0.2微米ptfe過濾器。將所需量的這種儲液添加到上文制成的嵌段共聚物溶液中并充分攪拌該混合物以形成均勻溶液,然后以2000rpm在ul-2涂覆基底上旋涂該配制的嵌段共聚物溶液30秒。在形成薄膜后,該涂覆硅片在140℃下烘烤5分鐘并立即冷卻至室溫。
進行sims(二次離子質譜學)分析獲得表19中所列的bcp+sap薄膜中的氟(f)深度分布。
表19.
氟在聚合物膜中的二次離子質譜學(sims)分析表明sap1和sap6系列都參與自組裝。氟曲線(f曲線)表明sap6系列添加劑具有比sap1高的在聚合物-空氣界面附近的分配。sap6-80比sap6-40高的分配表明可通過改變含f的結構部分的比率設計在嵌段共聚物結構域中具有不同分布狀況的saps。圖107-112分別是實施例227-232的濺射數據的圖。圖107-112分別是實施例227-232的濺射數據的圖。
本文所用的術語僅用于描述具體實施方案并且無意限制本發明。除非上下文清楚地另行指明,本文所用的單數形式“一”和“該”意在也包括復數形式。還要理解的是,術語“包含”和/或“含”在本說明書中使用時規定了所述特征、整數、步驟、操作、元件和/或組分的存在,但不排除一個或多個其它特征、整數、步驟、操作、元件、組分和/或它們的組合的存在或加入。當使用范圍以使用兩個數值界限x和y(例如xppm至yppm的濃度)表示可能的值時,除非另行指明,該值可以是x、y或在x和y之間的任何數值。
為舉例說明給出本發明的描述,但無意窮舉或限于所公開的形式的本發明。許多修改和變動是本領域普通技術人員顯而易見的而不背離本發明的范圍。選擇和描述實施方案以最好地解釋本發明的原理和它們的實際用途,并使本領域普通技術人員能夠理解本發明。
- 還沒有人留言評論。精彩留言會獲得點贊!