可交聯型多甲氧基側鏈二氟單體及其制備方法與流程
本發明屬于材料技術領域,具體涉及一類可交聯型多甲氧基側鏈二氟單體及其制備方法。
背景技術:
燃料電池作為一種新興的能源轉換裝置,具有轉化效率高、環境污染小等諸多優點,質子交換膜作為燃料電池的核心部件,其性能的優劣至關重要。目前,國內外有關磺化聚醚酮和磺化聚醚砜的制備和性能方面的研究報道非常多,磺化聚醚酮和磺化聚醚砜被認為是最有希望實用化的新型質子交換膜材料。磺化聚醚酮和磺化聚醚砜具有較高的質子傳導率,但存在電導率低、穩定性差的不足。
質子交換膜通過功能性單體化合物的聚合制備,按結構可分為主鏈型聚合物和側鏈型聚合物。通過調控單體化合物或聚合物的化學結構等手段,可以改善質子交換膜的性能。在分子鏈上引入側基是目前研究的一個重要研究方向,其目的是利用側基,局部破壞分子鏈的規整性,從而改善聚合物溶解性、加工性能,同時還可以賦予聚合物新的性能。與在主鏈上改性相比,在側鏈上改性的途徑比在主鏈上改性的途徑要多,而且效果也好。經過文獻調研,將親水的磺酸根通過側鏈連接在疏水聚合物的骨架上,能夠有效的促進磺酸根的聚集,形成寬暢的離子傳輸通道,提高質子交換膜的電導率。
近來有報道通過交聯反應來形成磺化交聯聚合物結構,提高磺化聚醚酮和磺化聚醚砜質子膜在高濕度下的力學性能、尺寸保持能力和抗氧化能力。發明人所在的研究小組前期授權的專利CN104262211A中公布了以1-(2,6-二氟苯基)-2-(3,5-二甲氧基苯基)乙烷-1,2-雙酮為側鏈二氟單體,通過與二酚、4,4’-二氟二苯基砜的縮聚反應制備出抗氧化性能強、質子傳導率高(80℃為171ms/cm,Nafion117 80℃下電
導率為165ms/cm)、可交聯的新型磺化聚醚砜質子交換膜材料。
技術實現要素:
本發明所要解決的技術問題在于提供一種可交聯型多甲氧基側鏈二氟單體,以及該單體的制備方法。
解決上述技術問題所采用的技術方案是該可交聯型多甲氧基側鏈二氟單體的結構式如下所示:
其中R1和R2代表H或且R1和R2不相同,X代表OCH3,n為1~3的整數。
本發明可交聯型多甲氧基側鏈二氟單體優選下述化合物A~K中的任意一種:
上述可交聯型多甲氧基側鏈二氟單體的合成路線為:
其中,R1和R2代表H或且R1和R2不相同,X代表OCH3,n為1~3的整數。
具體合成步驟如下:
1、制備化合物b和化合物c
以甲醇為溶劑,將化合物a與N-溴代丁二酰亞胺(NBS)、KBr在25~28℃下反應5~6小時,反應結束后,分離純化產物,制備成化合物b和化合物c。
2、制備單體
在氮氣保護下,將化合物b或化合物c與化合物d、碳酸鈉、四(三苯基)磷鈀、四丁基溴化銨按摩爾比為1:1.2~1.4:1.5~3:0.03~0.06:0.05~0.15,回流反應7~9小時,反應結束后,分離純化產物,制備成單體。
上述步驟1中,優選化合物a與N-溴代丁二酰亞胺、KBr的摩爾比為1:1.1:1.1。
上述步驟2中,優選化合物b或化合物c與化合物d、碳酸鈉、四(三苯基)磷鈀、四丁基溴化銨的摩爾比為1:1.2:2:0.05:0.1。
本發明的有益效果如下:
1、本發明單體含有可聚合二氟基團和可功能化的甲氧基,其經過縮聚反應和功能團轉化后可以得到磺化的聚合物,該功能化聚合物可作為燃料電池聚合物電解質膜材料。
2、本發明二氟單體擁有較長的側鏈,側鏈含多個可以功能化的甲氧基,更易形成離子化通道,保證了制備出的交換膜有較高的電導率。
3、本發明單體中含有偶酰基團,可以與氨基發生交聯,制備出網狀結構的聚合物,期望可以提高交換膜的機械性能。
附圖說明
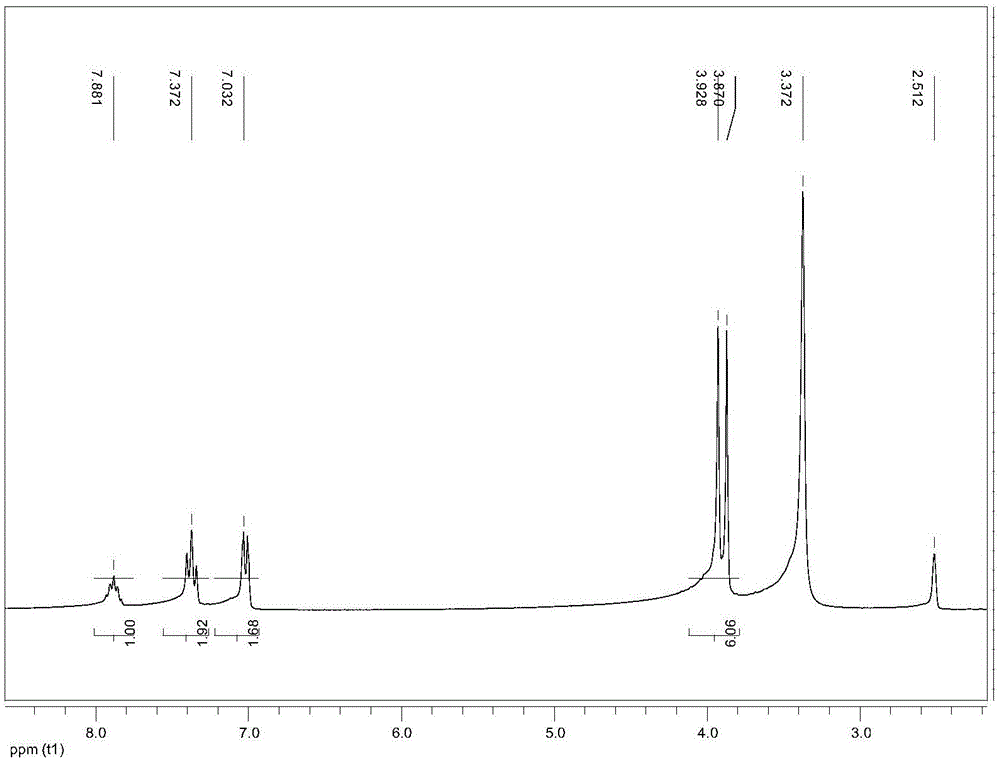
圖1是化合物b的核磁共振譜圖。
圖2是實施例1制備的可交聯型多甲氧基側鏈二氟單體的核磁共振譜圖。
圖3是實施例1制備的可交聯型多甲氧基側鏈二氟單體的紅外吸收譜圖。
圖4是實施例1制備的可交聯型多甲氧基側鏈二氟單體的質譜圖。
具體實施方式
下面結合附圖和實施例對本發明進一步詳細說明,但本發明的保護范圍不限于這些實施例。
實施例1
制備結構式如下的交聯型四甲氧基側鏈二氟單體(化合物J)
1、制備化合物b和化合物c
在250mL單口燒瓶中加入5.00g(16.33mmol)化合物a和100mL乙醇,待化合物a完全溶解后,依次加入2.14g(17.96mmol)KBr、3.19g(17.96mmol)N-溴代丁二酰亞胺,開啟磁力攪拌,室溫反應6小時。反應結束后,濃縮反應液,加入100mL去離子水在50℃下攪拌5小時。待完成后濾去水相得到淡黃色固體,以石油醚和乙酸乙酯體積比為2:1的混合液重結晶,得到淡黃色固體化合物b,其收率為53%,結構表征數據為:1H-NMR(DMSO-d6,300MHz,TMS)δ(ppm):3.870(s,3H),3.928(s,3H),7.007(s,1H),7.032(s,1H),7.372(t,2H),7.881(m,1H)(具體見圖1)。從重結晶的母液中柱層析分離得到化合物c,化合物c以副產物形式得到,其收率僅為8%。
2、制備單體
在氮氣保護下,向150mL三口燒瓶中加入4.00g(10.38mmol)化合物b、2.20g(20.77mmol)碳酸鈉、2.27g(12.46mmol)3,5-二甲氧基苯硼酸、0.60g(0.51mmol)四(三苯基)磷鈀、0.02g(0.14mmol)四丁基溴化銨和60mL甲苯、60mL蒸餾水、15mL乙二醇二甲醚,開啟磁力攪拌,加熱回流反應8小時。反應完成后,將反應液過濾,用二氯甲烷萃取,有機相經減壓濃縮,柱層析純化(展開劑為石油醚與乙酸乙酯體積比的10:1的混合液),柱層析后以石油醚與乙酸乙酯體積比5:1的混合液重結晶,得到白色色晶體化合物J,其收率為36%,結構表征數據為:1H-NMR(DMSO-d6,300MHz,TMS)δ(ppm):3.697(s,3H),3.802(s,6H),3.902(s,3H),6.271(s,1H),6.355(s,1H),6.600(s,1H),6.686(s,1H),6.933(s,1H),7.007(t,2H),7.415(t,1H)(具體見圖2)。
IR(KRr壓片,cm-1):3085,3016,2947,2944,1703,1616,1456,1371,1301,1209,1141,786(具體見圖3)。
MS m/z(rel.int.):441.92(M+,1.05),302.98(2.75),301.97(18.25),300.93(100),285.94(8.23),270.92(3.76),256.94(5.44),241.93(3.21),140.86(20.87),112.87(15.18)(具體見圖4)。
實施例2
制備結構式如下的可交聯型三甲氧基側鏈二氟單體(化合物A)
在實施例1的步驟1中,將化合物a和其他原料的用量均擴大10倍,其他條件與實施例1相同,制備成化合物c。然后按照實施例1的步驟2,將所用的3,5-二甲氧基苯硼酸用等摩爾的4-甲氧基苯硼酸替換,化合物b用等摩爾的化合物c替換,其他步驟與實施例1相同,制備成單體化合物A,其收率為66.7%。
實施例3
制備結構式如下的可交聯型三甲氧基側鏈二氟單體(化合物I)
在實施例1的步驟2中,所用的35-二甲基苯硼酸用等摩爾4-甲氧基苯硼酸替換,其他步驟與實施例1相同,制備成化合物I,其收率為53.8%。
實施例4
制備結構式如下的可交聯型五甲氧基側鏈二氟單體(化合物K)
在實施例1的步驟2中,所用的3,5-二甲基苯硼酸用等摩爾3,4,5-三甲氧基苯硼酸替換,其他步驟與實施例1相同,制備成化合物K,其收率為13.2%。
- 還沒有人留言評論。精彩留言會獲得點贊!