具有抗癌作用及抗幽門螺桿菌活性的化合物及其制造方法、以及其用途與流程
本申請為2014年2月24日提交的申請號為201480023381.3且發明名稱為“新化合物及其制造方法、以及其用途”的專利申請的分案申請。
本發明涉及具有抗癌作用及抗幽門螺桿菌活性的化合物、其制造方法、以及含有所述化合物的組合物、抗癌劑、和抗幽門螺桿菌(helicobacterpylori)劑。
背景技術:
癌組織以癌細胞和被稱為間質的周圍正常組織混在一起的形式構成。以下情況逐漸明確:所述間質由血管、細胞外基質、成纖維細胞樣細胞(也有時簡稱為“間質細胞”)等各種各樣的因子構成,并與癌的增殖密切相關。已知所述間質之中、特別是間質細胞介由粘接、分泌因子以正、負來控制癌細胞的增殖(例如參照非專利文獻1)。這種狀況下,進行更有用的新抗癌劑的探索,并強烈要求其的快速提供。
在胃潰瘍、十二指腸潰瘍等胃和十二指腸損害中,已知有由幽門螺桿菌(helicobacterpylori)引發的。于是,提出了喹啉酮化合物作為具有抗幽門螺桿菌活性的化合物(例如參照非專利文獻2和專利文獻1)。然而,不能說所述提出的喹啉酮化合物用作藥品是足夠的,要求新的具有抗幽門螺桿菌活性的化合物。
現有技術文獻
專利文獻
專利文獻1:美國專利第5942619號說明書
非專利文獻
非專利文獻1:kawada,m.,inoue,h.,masuda,t.,andikeda,d.insulin-likegrowthfactor-isecretedfromprostatestromalcellsmediatestumor-stromalcellinteractionsoftheprostatecancer.cancerres.66,4419-4425(2006).
非專利文獻2:dekker,k.a.,inagaki,t.,gootz,t.d.,huang,l.h.,kojimay.,kohlbrenner,w.e.,matsunaga,y.,mcguirk,p.r.,nomura,e.,sakakibara,t.sakemis.,suzuki,y.,yamauchi,y.,andkojima,n.newquinolonecompoundsfrompseudonocardiasp.withselectiveandpotentanti-helicobacterpyloriactivity:taxonomyofproducingstrain,fermentation,isolation,structuralelucidationandbiologicalactivities.j.antibiot.51,145-152(1998).
技術實現要素:
發明所要解決的課題
本發明是鑒于上述以往技術而進行的,其課題在于實現以下目的。即,本發明的目的在于,提供具有優異的抗癌作用、或優異的抗幽門螺桿菌活性的新化合物、和該新化合物的制造方法、以及利用所述新化合物的含化合物的組合物、抗癌劑、和抗幽門螺桿菌劑。
解決技術問題的手段
作為解決所述課題的手段,如下。即,
<1>一種化合物,其特征在于,以下述結構式(1)~(13)中的任一個表示。
其中,所述結構式(1)~(13)中,me表示甲基。
<2>下述結構式(3)、(4)、和(8)中的任一個表示的化合物的制造方法,其特征在于,使下述結構式(17)、(18)、和(19)中的任一個表示的化合物、與堿金屬的氫氧化物和堿土類金屬的氫氧化物中的至少一個進行反應。
<3>下述結構式(5)表示的化合物的制造方法,其特征在于,使下述結構式(21)表示的化合物、與堿金屬的醇鹽、堿金屬的碳酸鹽和堿金屬的氫化物中的至少一個反應后,使所述反應物與溴乙酸甲酯進行反應。
其中,所述結構式(5)中,me表示甲基。
<4>下述結構式(6)表示的化合物的制造方法,其特征在于,使下述結構式(5)表示的化合物、與堿金屬的氫氧化物和堿土類金屬的氫氧化物中的至少一個反應后,使所述反應物的ph為酸性。
其中,所述結構式(5)中,me表示甲基。
<5>下述結構式(1)表示的化合物的制造方法,其特征在于,使下述結構式(21)表示的化合物、與堿金屬的碳酸鹽和堿金屬的氫化物中的至少一個、與溴乙酸甲酯進行反應。
其中,所述結構式(1)中,me表示甲基。
<6>下述結構式(2)表示的化合物的制造方法,其特征在于,使下述結構式(1)表示的化合物、與堿金屬的氫氧化物和堿土類金屬的氫氧化物中的至少一個反應后,使所述反應物的ph為酸性。
其中,所述結構式(1)中,me表示甲基。
<7>下述結構式(7)表示的化合物的制造方法,其特征在于,使下述結構式(21)表示的化合物、與堿金屬的醇鹽和堿金屬的氫化物中的至少一個反應后,使所述反應物與溴化氰進行反應。
<8>下述結構式(9)表示的化合物的制造方法,其特征在于,使下述結構式(6)表示的化合物、與叔胺和吡啶類中的至少一個、與疊氮磷酸二苯酯、與甲硫醇鈉進行反應。
其中,所述結構式(9)中,me表示甲基。
<9>下述結構式(10)表示的化合物的制造方法,其特征在于,使下述結構式(6)表示的化合物、與叔胺和吡啶類中的至少一個、與疊氮磷酸二苯酯反應后,使所述反應物與甲硫醇鈉進行反應。
其中,所述結構式(10)中,me表示甲基。
<10>下述結構式(11)表示的化合物的制造方法,其特征在于,使下述結構式(20)表示的化合物、與堿金屬的醇鹽和堿金屬的氫化物中的至少一個反應后,使所述反應物與硫氰酸氯甲酯進行反應。
<11>下述結構式(12)表示的化合物的制造方法,其特征在于,在乙腈的存在下,使下述結構式(11)表示的化合物、與甲硫醇鈉進行反應。
其中,所述結構式(12)中,me表示甲基。
<12>下述結構式(13)表示的化合物的制造方法,其特征在于,在乙腈的存在下,使下述結構式(11)表示的化合物、與甲硫醇鈉反應后,使所述反應物與甲基化試劑進行反應。
其中,所述結構式(13)中,me表示甲基。
<13>一種含化合物的組合物,其特征在于,含有所述<1>所述的化合物。
<14>一種抗癌劑,其特征在于,含有所述<1>所述的化合物。
<15>一種抗幽門螺桿菌劑,其特征在于,含有所述<1>所述的化合物。
<16>一種用于預防或治療癌的方法,其特征在于,對個體給予所述<14>所述的抗癌劑。
<17>一種用于預防或治療由幽門螺桿菌帶來的感染病的方法,其特征在于,對個體給予所述<15>所述的抗幽門螺桿菌劑。
<18>一種用于預防或治療由幽門螺桿菌引起的胃和十二指腸損害的方法,其特征在于,對個體給予所述<15>所述的抗幽門螺桿菌劑。
發明效果
根據本發明,能夠實現所述目的,能夠提供具有優異的抗癌作用、或優異的抗幽門螺桿菌活性的新化合物、和該新化合物的制造方法、以及利用所述新化合物的含化合物的組合物、抗癌劑、和抗幽門螺桿菌劑。
附圖說明
圖1a:圖1a是表示試驗例2-1中的腫瘤體積的變化的圖表。
圖1b:圖1b是表示試驗例2-1中的腫瘤重量(從腫瘤接種起第21天)的圖表。
圖1c:圖1c是表示試驗例2-2中的腫瘤體積的變化的圖表。
圖1d:圖1d是表示試驗例2-2中的腫瘤重量(從腫瘤接種起第21天)的圖表。
具體實施方式
(新化合物)
本發明的化合物是下述結構式(1)~(13)表示的化合物,是本發明者們發現的新化合物。
其中,所述結構式(1)~(13)中,me表示甲基。
<結構式(1)表示的化合物的理化性質>
作為所述結構式(1)表示的化合物的理化性質,如下。
(1)外觀:無色油狀物
(2)分子式:c21h29o3n
(3)高分辨質譜分析(hresi-ms)(m/z):
實驗值344.2221(m+h)+
計算值344.2220(以c21h30o3n計)
(4)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:2953,2925,2854,1765,1618,1596,1437,1123,968,768,680
(5)質子核磁共振光譜(400mhz,cdcl3):
δ=0.87(3h,t,j=6.6),1.20-1.48(10h,m),1.71(2h,m),2.43(3h,s),2.95(2h,m),3.86(3h,s),4.62(2h,s),7.47(1h,ddd,j=8.2,6.8,1.1),7.62(1h,ddd,j=8.4,6.8,1.3),8.00(1h,d,j=8.4),8.05(1h,d,j=8.2)
(6)碳13核磁共振光譜(100mhz,cdcl3):
δ=11.90,14.08,22.63,28.99,29.23,29.49,29.86,31.83,37.02,52.32,70.26,120.85,120.73,121.39,121.76,125.73,128.82,128.85,147.84,159.03,164.32,168.95
<結構式(2)表示的化合物的理化性質>
作為所述結構式(2)表示的化合物的理化性質,如下。
(1)外觀:白色粉狀
(2)熔點:59℃~62℃
(3)分子式:c20h27o3n
(4)高分辨質譜分析(hresi-ms)(m/z):
實驗值330.2064(m+h)+
計算值330.2064(以c20h28o3n計)
(5)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:2927,2855,2713,1736,1642,1589,1227,1181,1078,764,724
(6)質子核磁共振光譜(600mhz,methanol-d4):
δ=0.88(3h,t,j=6.8),1.25-1.41(8h,m),1.50(2h,m),1.78(2h,m),2.54(3h,s),3.15(2h,t,m),4.92(2h,s),7.54(1h,ddd,j=8.2,7.2,1.0),7.92(1h,ddd,j=8.5,6.8,1.0),8.07(1h,brd,j=8.5),8.42(1h,brd,j=8.2)
(7)碳13核磁共振光譜(150mhz,methanol-d4):
δ=12.23,14.40,23.67,29.93,30.27,30.34,30.74,32.97,35.07,72.72,122.81,123.71,123.85,124.62,129.16,133.85,142.14,164.30,167.46,171.91
<結構式(3)表示的化合物的理化性質>
作為所述結構式(3)表示的化合物的理化性質,如下。
(1)外觀:白色粉狀
(2)熔點:178℃~181℃
(3)分子式:c19h25on
(4)高分辨質譜分析(hresi-ms)(m/z):
實驗值284.2011(m+h)+
計算值284.2009(以c19h26on計)
(5)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:3064,2957,2933,1670,1638,1614,1555,1500,1371,1358,1152,1028,998,967,756,691
(6)質子核磁共振光譜(600mhz,cdcl3):
δ=0.95(6h,d,j=6.5),1.44(2h,m),1.69(2h,m),2.01(2h,q,j=6.8),2.15(3h,s),2.21(1h,m),2.70(2h,m),5.27-5.32(1h,m),5.36--5.39(1h,m),7.28(1h,ddd,j=8.2,5.8,1.0),7.32(1h,brd,j=8.2),7.52(1h,ddd,j=8.2,5.5,1.4),8.36(1h,dd,j=8.2,1.4),8.65(1h,br)
(7)碳13核磁共振光譜(150mhz,cdcl3):
δ=10.65,22.64,27.77,29.22,30.98,32.09,32.96,115.72,116.69,123.00,123.66,126.14,126.30,131.25,138.49,138.78,148.54,178.16
<結構式(4)表示的化合物的理化性質>
作為所述結構式(4)表示的化合物的理化性質,如下。
(1)外觀:白色粉狀
(2)熔點:240℃~244℃;
(3)分子式:c14h17on
(4)高分辨質譜分析(hresi-ms)(m/z):
實驗值216.1385(m+h)+
計算值216.1383(以c14h18on計)
(5)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:3059,2956,1636,1609,1554,1505,1369,1359,1189,998,762,695
(6)質子核磁共振光譜(400mhz,dmso-d6):
δ=0.89(6h,d,j=6.6),1.94(3h,s),1.99(1h,m),2.53(2h,d,j=7.5),7.18(1h,ddd,j=8.2,6.6,1.4),7.46(1h,d,j=8.2),7.52(1h,ddd,j=8.2,6.6,1.4),8.01(1h,dd,j=8.2,1.1),11.23(1h,brs)
(7)碳13核磁共振光譜(100mhz,dmso-d6):
δ=11.03,22.28,28.30,114.68,117.74,122.39,123.00,125.18,131.08,139.33,148.76,176.43
<結構式(5)表示的化合物的理化性質>
作為所述結構式(5)表示的化合物的理化性質,如下。
(1)外觀:無色油狀物
(2)分子式:c21h29o3n
(3)高分辨質譜分析(hresi-ms)(m/z):
實驗值344.2222(m+h)+
計算值344.2220(以c21h30o3n計)
(4)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:2953,2922,1743,1635,1617,1558,1507,1214,994,760,688
(5)質子核磁共振光譜(400mhz,cdcl3):
δ=0.89(3h,t,j=6.6),1.21-1.51(10h,m),1.60(2h,m),2.22(3h,s),2.74(2h,br),3.80(3h,s),4.90(2h,s),7.20(1h,d,j=8.7),7.33(1h,ddd,j=8.0,6.8,0.7),7.58(1h,ddd,j=8.7,
(6)碳13核磁共振光譜(100mhz,cdcl3):
δ=11.65,14.06,22.60,28.06,29.13,29.17,29.75,30.93,31.75,48.47,53.01,114.19,117.55,123.11,124.79,127.30,131.89,140.66,150.66,168.69,177.39
<結構式(6)表示的化合物的理化性質>
作為所述結構式(6)表示的化合物的理化性質,如下。
(1)外觀:白色粉狀
(2)熔點:161℃~163℃
(3)分子式:c20h27o3n
(4)高分辨質譜分析(hresi-ms)(m/z):
實驗值330.2063(m+h)+
計算值330.2064(以c20h28o3n計)
(5)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:2955,2925,2853,1725,1635,1593,1506,1191,976,760,689
(6)質子核磁共振光譜(400mhz,cdcl3):
δ=0.87(3h,t,j=6.4),1.20-1.65(12h,m),2.19(3h,s),2.80(2h,br),4.98(2h,br),7.26(1h,t,j=8.0),7.42(1h,d,j=8.7),7.54(1h,ddd,j=8.7,6.8,1.1),8.39(1h,dd,j=8.0,1.1)
(7)碳13核磁共振光譜(100mhz,cdcl3):
δ=11.91,14.06,22.68,27.85,29.12,29.78,31.26,31.73,49.71,115.46,117.21,123.86,123.94,126.69,132.47,140.53,154.22,169.35,176.57
<結構式(7)表示的化合物的理化性質>
作為所述結構式(7)表示的化合物的理化性質,如下。
(1)外觀:無色油狀物
(2)分子式:c19h24on2
(3)高分辨質譜分析(hresi-ms)(m/z):
實驗值297.1961(m+h)+
計算值297.1961(以c19h25on2計)
(4)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:2961,2926,2853,2237,1628,1576,1470,1292,1191,761,693
(5)質子核磁共振光譜(400mhz,cdcl3):
δ=0.87(3h,t,j=6.8),1.20-1.50(10h,m),1.71(2h,m),2.15(3h,s),2.93(2h,m),7.47(1h,m),7.73(2h,m),8.33(1h,ddd,j=8.0,0.92,1.1)
(6)碳13核磁共振光譜(100mhz,cdcl3):
δ=11.20,14.05,22.59,28.00,29.07,29.11,29.42,31.73,31.80,106.42,116.28,120.30,123.35,126.23,127.08,133.34,137.25,146.10.177.31
<結構式(8)表示的化合物的理化性質>
作為所述結構式(8)表示的化合物的理化性質,如下。
(1)外觀:黃色粉狀
(2)熔點:>260℃
(3)分子式:c18h15on
(4)高分辨質譜分析(hresi-ms)(m/z):
實驗值284.1046(m+na)+
計算值284.1046(以c18h15onna計)
(5)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:3o64,2938,1628,1570,1507,1387,1359,1187,965,755,690
(6)質子核磁共振光譜(400mhz,dmso-d6):
δ=2.13(3h,s),7.21(1h,ddd,j=8.5,6.8,1.1),7.32-7.50(5h,m),7.55-7.59(1h,m),7.67-7.72(3h,m),8.03(1h,dd,j=8.2,1.4),11.20(1h,s)
(7)碳13核磁共振光譜(100mhz,dmso-d6):
δ=10.66,115.68,118.18,121.14,122.55,123.13,125.14,127.57,129.16,129.30,131.61,135.11,135.94,139.75,143.14,176.76
<結構式(9)表示的化合物的理化性質>
作為所述結構式(9)表示的化合物的理化性質,如下。
(1)外觀:無色油狀物
(2)分子式:c21h29o2ns
(3)高分辨質譜分析(hresi-ms)(m/z):
實驗值360.1994(m+h)+
計算值360.1992(以c21h30o2ns計)
(4)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:2924,2852,1687,1614,1594,1542,1193,1028,757,558
(5)質子核磁共振光譜(400mhz,cdcl3):
δ=0.88(3h,t,j=6.4),1.20-1.50(10h,m),1.60(2h,br),2.23(3h,s),2.33(3h,s),2.51-2.99(2h,br),5.01(2h,br),7.21(1h,d,j=8.7),7.34(1h,dd,j=8.0、6.6),7.58(1h,ddd,j=8.7,6.6,1.4),8.47(1h,dd,j=8.0,1.4)
(6)碳13核磁共振光譜(100mhz,cdcl3):
δ=11.38,11.68,14.06,22.60,28.16,29.13,29.16,29.75,31.03,31.74,55.95,114.61,117.94,123.32,127.29,131.97,132.02,140.73,150.65,177.49,196.82
<結構式(10)表示的化合物的理化性質>
作為所述結構式(10)表示的化合物的理化性質,如下。
(1)外觀:無色油狀物
(2)分子式:c21h30o2n2s
(3)高分辨質譜分析(hresi-ms)(m/z):
實驗值397.1921(m+na)+
計算值397.1920(以c21h30o2n2nas計)
(4)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:3169,2955,2927,1671,1615,1595,1556,1492,1195,1084,760,651
(5)質子核磁共振光譜(600mhz,cdcl3):
δ=0.89(3h,t,j=6.7),1.22-1.45(15h,m),2.45(2h,br),2.46(3h,s),5.67(2h,br),7.24(1h,ddd,j=7.9,6.8,1.0),7.49(1h,d,j=8.6),7.59(1h,ddd,j=8.6,6.8,1.4),8.26(1h,dd,j=7.9,1.4),8.78(1h,br)
(6)碳13核磁共振光譜(150mhz,cdcl3):
δ=11.05,12.22,14.02,22.59,28.48,29.11,29.16,29.73,30.73,31.77,52.58,115.46,117.00,123.24,124.37,126.89,132.32,139.56,151.60,168.61,177.27
<結構式(11)表示的化合物的理化性質>
作為所述結構式(11)表示的化合物的理化性質,如下。
(1)外觀:黃色油狀物
(2)分子式:c19h24on2s
(3)高分辨質譜分析(hresi-ms)(m/z):
實驗值329.1682(m+h)+
計算值329.1682(以c19h25on2s計)
(4)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:2961,2926,2853,2237,1628,1576,1470,1292,1191,987,761,693
(5)質子核磁共振光譜(400mhz,cdcl3):
δ=0.90(3h,t,j=6.6),1.23-1.71(10h,m),2.19(3h,s),2.84(2h,m),5.71(2h,s),7.38(1h,ddd,j=8.0,6.8,0.9),7.46(1h,d,j=8.7),7.68(1h,ddd,j=8.7,6.8,1.6),8.45(1h,dd,j=8.0,1.6)
(6)碳13核磁共振光譜(100mhz,cdcl3):
δ=11.60,14.05,22.56,28.59,28.88,29.76,30.55,31.66,56.26,114.42,118.19,123.83,124.60,127.31,132.41,139.86,141.68,149.60,177.64
<結構式(12)表示的化合物的理化性質>
作為所述結構式(12)表示的化合物的理化性質,如下。
(1)外觀:黃色粉狀
(2)熔點:167℃~170℃
(3)分子式:c20h28on2s2
(4)高分辨質譜分析(hresi-ms)(m/z):
實驗值399.1534(m+na)+
計算值399.1535(以c20h28on2nas2計)
(5)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:3119,2958,2918,2850,1619,1598,1538,1282,1199,1105,938,764,688
(6)質子核磁共振光譜(400mhz,cdcl3):
δ=0.86(3h,t,j=6.6),1.21-1.50(13h,m),2.22-2.58(2h,br),2.76(3h,s),5.68-6.41(2h,br),7.22(1h,t,j=7.8),7.44(1h,d,j=8.7),7.59(1h,ddd,j=8.7,7.8,1.1),8.15(1h,d,j=7.8),10.09(1h,br)
(7)碳13核磁共振光譜(100mhz,cdcl3):
δ=10.74,14.02,18.01,22.53,28.29,28.77,29.64,30.85,31.59,58.21,115.93,116.84,123.54,123.91,126.35,132.65,139.39,152.23.177.13.199.84
<結構式(13)表示的化合物的理化性質>
作為所述結構式(13)表示的化合物的理化性質,如下。
(1)外觀:白色粉狀
(2)熔點:92℃~94℃
(3)分子式:c21h30on2s2
(4)高分辨質譜分析(hresi-ms)(m/z):
實驗值413.1689(m+na)+
計算值413.1692(以c21h30on2nas2計)
(5)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:2958,2922,2852,1618,1595,1566,1492,1370,1277,1192,1004,769,700
(6)質子核磁共振光譜(400mhz,cdcl3):
δ=0.89(3h,t,j=6.8),1.24-1.50(8h,m),1.64(2h,m),2.22(3h,s),2.28(3h,s),2.71(3h,s),2.77(2h,m),5.60(2h,s),7.31(2h,m),7.55(1h,ddd,j=8.4,7.1,1.6),8.46(1h,dd,j=8.0,1.6)
(7)碳13核磁共振光譜(100mhz,cdcl3):
δ=11.50,14.06,14.75,15.03,22.61,28.28,28.92,29.83,30.66,31.76,63.75,115.84,116.97,122.77,124.81,126.75,131.27,141.06,151.35,161.39,177.54
所述化合物是否具有以所述結構式(1)~(13)中的任一個表示的結構能夠通過適當選擇的各種分析方法進行確認,例如可以舉出所述質譜分析法、所述紅外分光法、所述質子核磁共振分光法、所述碳13核磁共振分光法、紫外分光法等分析方法等。應予說明,由所述各分析方法而得的測定值有時會產生一些誤差,但只要是本領域技術人員,就可以容易地鑒定出所述化合物具有以所述結構式(1)~(13)中的任一個表示的結構。
所述化合物可以是以所述結構式(1)~(13)中的任一個表示的化合物的鹽。
作為所述鹽,只要是可以在藥理學上容許的鹽,就沒有特別限制,能夠根據目的適當選擇,例如可以舉出乙酸鹽、檸檬酸鹽等有機鹽、鹽酸鹽、碳酸鹽等。
所述結構式(1)~(13)中的任一個表示的化合物可以為其互變異構體。
作為所述結構式(1)~(13)中的任一個表示的化合物的制造方法,沒有特別限制,能夠根據目的適當選擇,但優選通過后述的本發明的制造方法制得。
<用途>
所述結構式(1)~(13)中的任一個表示的化合物具有優異的抗癌作用、或優異的抗幽門螺桿菌活性,是安全性高的化合物。因此,所述結構式(1)~(13)中的任一個表示的化合物例如可以優選用作后述的含本發明的化合物的組合物、本發明的抗癌劑、本發明的抗幽門螺桿菌劑等的有效成分。
(化合物的制造方法)
<所述結構式(3)、(4)、和(8)中的任一個表示的化合物的制造方法>
作為所述結構式(3)、(4)、和(8)中的任一個表示的化合物的制造方法,只要是使下述結構式(17)、(18)、和(19)中的任一個表示的化合物、與堿金屬的氫氧化物和堿土類金屬的氫氧化物中的至少一個進行反應的方法,就沒有特別限制,能夠根據目的適當選擇。
所謂所述堿金屬是指鋰、鈉、鉀、銣、銫、鈁。所謂所述堿土類金屬,是指鈣、鍶、鋇、鐳。
作為所述堿金屬的氫氧化物,沒有特別限制,能夠根據目的適當選擇,例如可以舉出氫氧化鋰、氫氧化鈉、氫氧化鉀等。
作為所述堿土類金屬的氫氧化物,沒有特別限制,能夠根據目的適當選擇,例如可以舉出氫氧化鋇等。
所述堿金屬的氫氧化物和所述堿土類的氫氧化物可以使用任一個,也可以將兩者并用。
所述堿金屬的氫氧化物可以單獨使用一種,也可以將兩種以上并用。另外,所述堿土類金屬的氫氧化物可以單獨使用一種,也可以將兩種以上并用。
所述堿金屬的氫氧化物和所述堿土類金屬的氫氧化物之中,優選氫氧化鈉。
下面,作為所述結構式(3)、(4)、和(8)中的任一個表示的化合物的制造方法的優選方式,將以氨基芐腈為起始物質的方式說明如下。
--結構式(14)表示的化合物的制造--
所述結構式(14)表示的化合物例如能夠按以下方式制造。
氬氣氛下使氨基芐腈溶解在無水四氫呋喃(以下有時稱為“thf”)中,冰浴下滴加溴化乙基鎂。在室溫下使其攪拌12小時后,在冰浴下滴加10%鹽酸水溶液。滴加完成后,在冰浴下加入氫氧化鈉,將ph調到7。分離有機層,用二乙基醚萃取水層。合并有機層進行芒硝干燥,蒸餾除去溶劑。能夠采用硅膠色層分離法(己烷:乙酸乙酯=6:1)純化得到的殘渣而獲得結構式(14)表示的化合物。
--結構式(17)表示的化合物的制造--
所述結構式(17)表示的化合物例如能夠按以下方式制造。
氬氣氛下使所述結構式(14)表示的化合物溶解在二氯甲烷中,加入三乙基胺。進一步在冰浴下滴加作為酰氯的反-8-甲基-6-壬酰氯,在室溫下攪拌。用0.1n鹽酸停止反應,用二氯甲烷萃取,接著用飽和碳酸氫鈉水溶液、食鹽水洗滌。將合并的有機層進行芒硝干燥后,蒸餾除去溶劑。能夠采用硅膠色層分離法(己烷:乙酸乙酯)純化得到的殘渣而獲得結構式(17)表示的化合物。
-結構式(3)表示的化合物的制造-
以所述結構式(3)表示的化合物例如能夠按以下方式制造。
在所述結構式(17)表示的化合物的二噁烷0.1m溶液中加入氫氧化鈉,在110℃下攪拌1小時~2小時。將反應溶液恢復到室溫,加入水,進一步加入1n鹽酸直至ph變成7。進一步加入己烷施加超聲波,則固體析出。抽濾固體,用水、己烷或己烷/乙酸乙酯=1:1的混合溶劑洗滌。能夠洗滌后使其干燥,從而獲得結構式(3)表示的化合物。
--結構式(18)表示的化合物的制造--
所述結構式(18)表示的化合物例如能夠按以下方式制造。
氬氣氛下使所述結構式(14)表示的化合物溶解在二氯甲烷中,加入三乙基胺。進一步在冰浴下滴加作為酰氯的異戊酰氯,在室溫下攪拌。用0.1n鹽酸停止反應,用二氯甲烷萃取,接著用飽和碳酸氫鈉水溶液、食鹽水洗滌。將合并的有機層進行芒硝干燥后,蒸餾除去溶劑。能夠采用硅膠色層分離法(己烷:乙酸乙酯)純化得到的殘渣而獲得結構式(18)表示的化合物。
-結構式(4)表示的化合物的制造-
所述結構式(4)表示的化合物例如能夠按以下方式制造。
在所述結構式(18)表示的化合物的1,4-二噁烷0.1m溶液中加入氫氧化鈉,在110℃下攪拌1小時~2小時。將反應溶液恢復到室溫,加入水,進一步加入1n鹽酸直至ph變成7。進一步加入己烷施加超聲波,則固體析出。抽濾固體,用水、己烷或己烷/乙酸乙酯=1:1的混合溶劑洗滌。能夠洗滌后使其干燥,從而獲得結構式(4)表示的化合物。
--結構式(19)表示的化合物的制造--
所述結構式(19)表示的化合物例如能夠按以下方式制造。
氬氣氛下使所述結構式(14)表示的化合物溶解在二氯甲烷中,加入三乙基胺。進一步在冰浴下滴加作為酰氯的肉桂酰氯,在室溫下攪拌。用0.1n鹽酸停止反應,用二氯甲烷萃取,接著用飽和碳酸氫鈉水溶液、食鹽水洗滌。將合并的有機層進行芒硝干燥后,蒸餾除去溶劑。能夠采用硅膠色層分離法(己烷:乙酸乙酯)純化得到的殘渣而獲得結構式(19)表示的化合物。
-結構式(8)表示的化合物的制造-
所述結構式(8)表示的化合物例如能夠按以下方式制造。
在所述結構式(19)表示的化合物的1,4-二噁烷0.1m溶液中加入氫氧化鈉,在110℃下攪拌1小時~2小時。將反應溶液恢復到室溫,加入水,進一步加入1n鹽酸直至ph變成7。進一步加入己烷施加超聲波,則固體析出。抽濾固體,用水、己烷或己烷/乙酸乙酯=1:1的混合溶劑洗滌。能夠洗滌后使其干燥,從而獲得結構式(8)表示的化合物。
<所述結構式(5)表示的化合物的制造方法>
作為所述結構式(5)表示的化合物的制造方法,只要是使下述結構式(21)表示的化合物、與堿金屬的醇鹽、堿金屬的碳酸鹽、和堿金屬的氫化物中的至少一個反應后、使所述反應物與溴乙酸甲酯進行反應的方法,就沒有特別限制,能夠根據目的適當選擇。
其中,所述結構式(5)中,me表示甲基。
所謂所述堿金屬,是指鋰、鈉、鉀、銣、銫、鈁。
作為所述堿金屬的醇鹽,沒有特別限制,能夠根據目的適當選擇,例如可以舉出叔丁醇鋰等。
作為所述堿金屬的碳酸鹽,沒有特別限制,能夠根據目的適當選擇,例如可以舉出碳酸鈉、碳酸鉀、碳酸氫鈉、碳酸氫鉀等。
作為所述堿金屬的氫化物,沒有特別限制,能夠根據目的適當選擇,例如可以舉出氫化鈉。
所述堿金屬的醇鹽、所述堿金屬的碳酸鹽、和所述堿金屬的氫化物可以單獨使用任一種,也可以將兩種以上并用。
所述堿金屬的醇鹽可以單獨使用一種,也可以將兩種以上并用。所述堿金屬的碳酸鹽可以單獨使用一種,也可以將兩種以上并用。另外,所述堿金屬的氫化物可以單獨使用一種,也可以將兩種以上并用。
所述堿金屬的醇鹽、所述堿金屬的碳酸鹽、和所述堿金屬的氫化物之中,優選叔丁醇鋰、碳酸鉀、氫化鈉,更優選叔丁醇鋰。
下面,作為以所述結構式(5)表示的化合物的制造方法的優選方式,將所述結構式(14)表示的化合物為起始物質的方式說明如下。
應予說明,所述結構式(14)表示的化合物能夠優選根據上述方法制造。
--結構式(16)表示的化合物的制造--
所述結構式(16)表示的化合物例如能夠按以下方式制造。
氬氣氛下使所述結構式(14)表示的化合物溶解在二氯甲烷中,加入三乙基胺。進一步在冰浴下滴加作為酰氯的壬酰氯,在室溫下攪拌。用0.1n鹽酸停止反應,用二氯甲烷萃取,接著用飽和碳酸氫鈉水溶液、食鹽水洗滌。將合并的有機層進行芒硝干燥后,蒸餾除去溶劑。能夠采用硅膠色層分離法(己烷:乙酸乙酯)純化得到的殘渣而獲得結構式(16)表示的化合物。
--結構式(21)表示的化合物的制造--
所述結構式(21)表示的化合物例如能夠按以下方式制造。
在所述結構式(16)表示的化合物的1,4-二噁烷0.1m溶液中加入氫氧化鈉,在110℃下攪拌1小時~2小時。將反應溶液恢復到室溫,加入水,進一步加入1n鹽酸直至ph變成7。進一步加入己烷施加超聲波,則固體析出。抽濾固體,用水、己烷或己烷/乙酸乙酯=1:1的混合溶劑洗滌。能夠洗滌后使其干燥,從而獲得結構式(21)表示的化合物。
-結構式(5)表示的化合物的制造-
所述結構式(5)表示的化合物例如能夠按以下方式制造。
氬氣氛下使所述結構式(21)表示的化合物溶解在thf中,加入叔丁醇鋰的thf溶液在室溫下攪拌20分鐘。接著,加入溴乙酸甲酯,進一步回流下攪拌12小時。加入水,使反應停止,用乙酸乙酯萃取。對有機層進行芒硝干燥,蒸餾除去溶劑。能夠將得到的殘渣采用硅膠色層分離法(己烷:乙酸乙酯=2:1)純化而獲得結構式(5)表示的化合物。
<所述結構式(6)表示的化合物的制造方法>
作為所述結構式(6)表示的化合物的制造方法,只要是使下述結構式(5)表示的化合物、與堿金屬的氫氧化物和堿土類金屬的氫氧化物中的至少一個反應后、使所述反應物的ph成酸性的方法,就沒有特別限制,能夠根據目的適當選擇。
其中,所述結構式(5)中,me表示甲基。
所謂所述堿金屬,是指鋰、鈉、鉀、銣、銫、鈁。所謂所述堿土類金屬,是指鈣、鍶、鋇、鐳。
作為所述堿金屬的氫氧化物,沒有特別限制,能夠根據目的適當選擇,例如可以舉出氫氧化鋰、氫氧化鈉、氫氧化鉀等。
作為所述堿土類金屬的氫氧化物,沒有特別限制,能夠根據目的適當選擇,例如可以舉出氫氧化鋇等。
所述堿金屬的氫氧化物和所述堿土類的氫氧化物可以使用任一個,也可以將兩者并用。
所述堿金屬的氫氧化物可以單獨使用一種,也可以將兩種以上并用。另外,所述堿土類金屬的氫氧化物可以單獨使用一種,也可以將兩種以上并用。
所述堿金屬的氫氧化物和所述堿土類金屬的氫氧化物之中,優選氫氧化鈉。
下面,作為所述結構式(6)表示的化合物的制造方法的優選方式,將以所述結構式(5)表示的化合物為起始物質的方式說明如下。
應予說明,所述結構式(5)表示的化合物能夠優選根據上述方法制造。
-結構式(6)表示的化合物的制造-
所述結構式(6)表示的化合物例如能夠按以下方式制造。
使所述結構式(5)表示的化合物溶解在乙醇(以下有時稱為“etoh”)與thf的混合溶劑中,加入2m氫氧化鈉水溶液,在室溫下攪拌2小時。冰浴下加入1n鹽酸將ph調到4,用乙酸乙酯萃取。能夠對有機層進行芒硝干燥,蒸餾除去溶劑,從而獲得結構式(6)表示的化合物。
<所述結構式(1)表示的化合物的制造方法>
作為所述結構式(1)表示的化合物的制造方法,只要是使下述結構式(21)表示的化合物、與堿金屬的碳酸鹽和堿金屬的氫化物中的至少一個、與溴乙酸甲酯進行反應的方法,就沒有特別限制,能夠根據目的適當選擇。
其中,所述結構式(1)中,me表示甲基。
所謂所述堿金屬,是指鋰、鈉、鉀、銣、銫、鈁。
作為所述堿金屬的碳酸鹽,沒有特別限制,能夠根據目的適當選擇,例如可以舉出碳酸鈉、碳酸鉀、碳酸氫鈉、碳酸氫鉀等。
作為所述堿金屬的氫化物,沒有特別限制,能夠根據目的適當選擇,例如可以舉出氫化鈉。
所述堿金屬的碳酸鹽和所述堿金屬的氫化物可以使用任一個,也可以將兩者并用。
所述堿金屬的碳酸鹽可以單獨使用一種,也可以將兩種以上并用。另外,所述堿金屬的氫化物可以單獨使用一種,也可以將兩種以上并用。
所述堿金屬的碳酸鹽和所述堿金屬的氫化物之中,優選碳酸鉀、氫化鈉,更優選碳酸鉀。
下面,作為所述結構式(1)表示的化合物的制造方法的優選方式,將以所述結構式(21)表示的化合物為起始物質的方式說明如下。
應予說明,所述結構式(21)表示的化合物能夠優選根據上述方法制造。
-結構式(1)表示的化合物的制造-
所述結構式(1)表示的化合物例如能夠按以下方式制造。
使所述結構式(21)溶解在n,n-二甲基甲酰胺(以下,有時稱為“dmf”)中,加入碳酸鉀、溴乙酸甲酯,在80℃下攪拌12小時。加入水,使反應停止,用乙酸乙酯萃取。對有機層進行芒硝干燥,蒸餾除去溶劑。能夠將得到的殘渣采用硅膠色層分離法(己烷:乙酸乙酯=2:1)純化而獲得結構式(1)表示的化合物。
<所述結構式(2)表示的化合物的制造方法>
作為所述結構式(2)表示的化合物的制造方法,只要是使下述結構式(1)表示的化合物、與堿金屬的氫氧化物和堿土類金屬的氫氧化物中的至少一個反應后、使所述反應物的ph成酸性的方法,就沒有特別限制,能夠根據目的適當選擇。
其中,所述結構式(1)中,me表示甲基。
所謂所述堿金屬,是指鋰、鈉、鉀、銣、銫、鈁。所謂所述堿土類金屬,是指鈣、鍶、鋇、鐳。
作為所述堿金屬的氫氧化物,沒有特別限制,能夠根據目的適當選擇,例如可以舉出氫氧化鋰、氫氧化鈉、氫氧化鉀等。
作為所述堿土類金屬的氫氧化物,沒有特別限制,能夠根據目的適當選擇,例如可以舉出氫氧化鋇等。
所述堿金屬的氫氧化物和所述堿土類的氫氧化物可以使用任一個,也可以將兩者并用。
所述堿金屬的氫氧化物可以單獨使用一種,也可以將兩種以上并用。另外,所述堿土類金屬的氫氧化物可以單獨使用一種,也可以將兩種以上并用。
所述堿金屬的氫氧化物和所述堿土類金屬的氫氧化物之中,優選氫氧化鈉。
作為使所述反應物的ph成酸性的方法,沒有特別限制,能夠根據目的適當選擇,例如可以舉出1n鹽酸等。
下面,作為所述結構式(2)表示的化合物的制造方法的優選方式,將以所述結構式(1)表示的化合物為起始物質的方式說明如下。
應予說明,所述結構式(1)表示的化合物能夠優選根據上述方法制造。
-結構式(2)表示的化合物的制造-
所述結構式(2)表示的化合物例如能夠按以下方式制造。
使所述結構式(1)表示的化合物溶解在etoh與thf的混合溶劑中,加入2m氫氧化鈉水溶液,在室溫下攪拌2小時。冰浴下加入1n鹽酸,將ph調到4,用乙酸乙酯萃取。能夠對有機層進行芒硝干燥,蒸餾除去溶劑,從而獲得結構式(2)表示的化合物。
<所述結構式(7)表示的化合物的制造方法>
作為所述結構式(7)表示的化合物的制造方法,只要是使下述結構式(21)表示的化合物、與堿金屬的醇鹽和堿金屬的氫化物中的至少一個反應后、使所述反應物與溴化氰進行反應的方法,就沒有特別限制,能夠根據目的適當選擇。
所謂所述堿金屬,是指鋰、鈉、鉀、銣、銫、鈁。
作為所述堿金屬的醇鹽,沒有特別限制,能夠根據目的適當選擇,例如可以舉出叔丁醇鋰等。
作為所述堿金屬的氫化物,沒有特別限制,能夠根據目的適當選擇,例如可以舉出氫化鈉。
所述堿金屬的醇鹽和所述堿金屬的氫化物可以使用任一個,也可以將兩者并用。
所述堿金屬的醇鹽可以單獨使用一種,也可以將兩種以上并用。另外,所述堿金屬的氫化物可以單獨使用一種,也可以將兩種以上并用。
所述堿金屬的醇鹽和所述堿金屬的氫化物之中,優選叔丁醇鋰、氫化鈉,更優選叔丁醇鋰。
下面,作為所述結構式(7)表示的化合物的制造方法的優選方式,將所述結構式(21)表示的化合物為起始物質的方式說明如下。
應予說明,所述結構式(21)表示的化合物能夠優選根據上述方法制造。
-結構式(7)表示的化合物的制造-
所述結構式(7)表示的化合物例如能夠按以下方式制造。
使所述結構式(21)表示的化合物溶解在thf中,加入叔丁醇鋰,在室溫下攪拌20分鐘。接著,加入溴化氰,在室溫下攪拌2小時。加入水,使反應停止,用乙酸乙酯萃取。對有機層進行芒硝干燥,蒸餾除去溶劑。能夠將得到的殘渣采用硅膠色層分離法(己烷:乙酸乙酯=2:1)純化,從而獲得結構式(7)表示的化合物(34.0mg,62%)。
<所述結構式(9)表示的化合物的制造方法>
作為所述結構式(9)表示的化合物的制造方法,只要是使下述結構式(6)表示的化合物、與叔胺和吡啶類中的至少一個、與疊氮磷酸二苯酯、與甲硫醇鈉進行反應的方法,就沒有特別限制,能夠根據目的適當選擇。
其中,所述結構式(9)中、me表示甲基。
作為所述叔胺,沒有特別限制,能夠根據目的適當選擇,例如可以舉出三乙基胺、n,n-二異丙基乙胺等。
作為所述吡啶類,沒有特別限制,能夠根據目的適當選擇,例如可以舉出吡啶、二甲基氨基吡啶等。
所述叔胺和所述吡啶類可以使用任一個,也可以將兩者并用。
所述叔胺可以單獨使用一種,也可以將兩種以上并用。另外,所述吡啶類可以單獨使用一種,也可以將兩種以上并用。
所述叔胺和所述吡啶類之中,優選三乙基胺。
下面,作為所述結構式(9)表示的化合物的制造方法的優選方式,將以所述結構式(6)表示的化合物為起始物質的方式說明如下。
應予說明,所述結構式(6)表示的化合物能夠優選根據上述方法制造。
-結構式(9)表示的化合物的制造-
所述結構式(9)表示的化合物例如能夠按以下方式制造。
使所述結構式(6)表示的化合物懸浮于thf中,加入三乙基胺、疊氮磷酸二苯酯(以下有時稱為“dppa”)、甲硫醇鈉,回流下攪拌2小時。加入氯化銨水溶液,用乙酸乙酯萃取。對有機層進行芒硝干燥,蒸餾除去溶劑。能夠將得到的殘渣采用硅膠色層分離法(己烷:乙酸乙酯=3:1)純化而獲得結構式(9)表示的化合物。
<所述結構式(10)表示的化合物的制造方法>
作為所述結構式(10)表示的化合物的制造方法,只要是使下述結構式(6)表示的化合物、與叔胺和吡啶類中的至少一個、與疊氮磷酸二苯酯反應后、使所述反應物與甲硫醇鈉進行反應的方法,就沒有特別限制,能夠根據目的適當選擇。
其中,所述結構式(10)中,me表示甲基。
作為所述叔胺,沒有特別限制,能夠根據目的適當選擇,例如可以舉出三乙基胺、n,n-二異丙基乙胺等。
作為所述吡啶類,沒有特別限制,能夠根據目的適當選擇,例如可以舉出吡啶、二甲基氨基吡啶等。
所述叔胺和所述吡啶類可以使用任一個,也可以將兩者并用。
所述叔胺可以單獨使用一種,也可以將兩種以上并用。另外,所述吡啶類可以單獨使用一種,也可以將兩種以上并用。
所述叔胺和所述吡啶類之中,優選三乙基胺。
下面,作為所述結構式(10)表示的化合物的制造方法的優選方式,將以所述結構式(6)表示的化合物為起始物質的方式說明如下。
應予說明,所述結構式(6)表示的化合物能夠優選根據上述方法制造。
-結構式(10)表示的化合物的制造-
所述結構式(10)表示的化合物例如能夠按以下方式制造。
使所述結構式(6)表示的化合物懸浮在thf中,在冰冷卻下加入三乙基胺、dppa,進一步回流下攪拌1小時。在其中加入甲硫醇鈉,進一步攪拌1小時。加入氯化銨水溶液,用乙酸乙酯萃取。對有機層進行芒硝干燥,蒸餾除去溶劑。能夠將得到的殘渣采用硅膠色層分離法(己烷:乙酸乙酯=5:1)純化而獲得結構式(10)表示的化合物。
<所述結構式(11)表示的化合物的制造方法>
作為所述結構式(11)表示的化合物的制造方法,只要是使下述結構式(20)表示的化合物、與堿金屬的醇鹽和堿金屬的氫化物中的至少一個反應后、使所述反應物與硫氰酸氯甲酯進行反應的方法,就沒有特別限制,能夠根據目的適當選擇。
所謂所述堿金屬,是指鋰、鈉、鉀、銣、銫、鈁。
作為所述堿金屬的醇鹽,沒有特別限制,能夠根據目的適當選擇,例如可以舉出叔丁醇鋰等。
作為所述堿金屬的氫化物,沒有特別限制,能夠根據目的適當選擇,例如可以舉出氫化鈉。
所述堿金屬的醇鹽和所述堿金屬的氫化物可以使用任一個,也可以將兩者并用。
所述堿金屬的醇鹽可以單獨使用一種,也可以將兩種以上并用。另外,所述堿金屬的氫化物可以單獨使用一種,也可以將兩種以上并用。
所述堿金屬的醇鹽和所述堿金屬的氫化物之中,優選叔丁醇鋰、氫化鈉,更優選叔丁醇鋰。
下面,作為所述結構式(11)表示的化合物的制造方法的優選方式,將以所述結構式(14)表示的化合物為起始物質的方式說明如下。
應予說明,所述結構式(14)表示的化合物能夠優選根據上述方法制造。
--結構式(15)表示的化合物的制造--
所述結構式(15)表示的化合物例如能夠按以下方式制造。
氬氣氛下使所述結構式(14)表示的化合物溶解在二氯甲烷中,加入三乙基胺。進一步在冰浴下滴加作為酰氯的辛酰氯,在室溫下攪拌。用0.1n鹽酸停止反應,用二氯甲烷萃取,接著用飽和碳酸氫鈉水溶液、食鹽水洗滌。將合并的有機層進行芒硝干燥后,蒸餾除去溶劑。能夠采用硅膠色層分離法(己烷:乙酸乙酯)純化得到的殘渣而獲得結構式(15)表示的化合物。
--結構式(20)表示的化合物的制造--
所述結構式(20)表示的化合物例如能夠按以下方式制造。
在所述結構式(15)表示的化合物的二噁烷0.1m溶液中加入氫氧化鈉,在110℃下攪拌1小時~2小時。將反應溶液恢復到室溫,加入水,進一步加入1n鹽酸直至ph變成7。進一步加入己烷施加超聲波,則固體析出。抽濾固體,用水、己烷或己烷/乙酸乙酯=1:1的混合溶劑洗滌。能夠洗滌后使其干燥,從而獲得結構式(20)表示的化合物。
-結構式(11)表示的化合物的制造-
所述結構式(11)表示的化合物例如能夠按以下方式制造。
氬氣氛下使所述結構式(20)表示的化合物溶解在thf中,加入叔丁醇鋰的thf溶液,在室溫下攪拌20分鐘。冰冷卻下在其中滴加硫氰酸氯甲酯,進一步在室溫下攪拌2小時。加入食鹽水使反應停止,用乙酸乙酯萃取。能夠對有機層進行芒硝干燥后,采用硅膠色層分離法(己烷:乙酸乙酯=2:1)純化得到的殘渣,從而獲得結構式(11)表示的化合物。
<所述結構式(12)表示的化合物的制造方法>
作為所述結構式(12)表示的化合物的制造方法,只要是在乙腈的存在下使下述結構式(11)表示的化合物與甲硫醇鈉進行反應的方法,就沒有特別限制,能夠根據目的適當選擇。
其中,所述結構式(12)中,me表示甲基。
下面,作為所述結構式(12)表示的化合物的制造方法的優選方式,將以所述結構式(11)表示的化合物為起始物質的方式說明如下。
應予說明,所述結構式(11)表示的化合物能夠優選根據上述方法制造。
-結構式(12)表示的化合物的制造-
所述結構式(12)表示的化合物例如能夠按以下方式制造。
在所述結構式(11)表示的化合物與甲硫醇鈉的混合物中加入乙腈,在室溫下攪拌20分鐘。加入飽和碳酸氫鈉水溶液,用乙酸乙酯萃取。能夠對有機層進行芒硝干燥后,采用硅膠色層分離法(己烷:乙酸乙酯=2:1)純化得到的殘渣,從而獲得結構式(12)表示的化合物。
<所述結構式(13)表示的化合物的制造方法>
作為所述結構式(13)表示的化合物的制造方法,只要是在乙腈的存在下使下述結構式(11)表示的化合物與甲硫醇鈉反應后、使所述反應物與甲基化試劑進行反應的方法,就沒有特別限制,能夠根據目的適當選擇。
其中,所述結構式(13)中,me表示甲基。
作為所述甲基化試劑,沒有特別限制,能夠根據目的適當選擇,例如可以舉出碘代甲烷、三氟甲磺酸甲酯、硫酸二甲酯、meerwein試劑等。所述甲基化試劑可以單獨使用一種,也可以將兩種以上并用。這些之中,優選碘代甲烷。
下面,作為所述結構式(13)表示的化合物的制造方法的優選方式,將以所述結構式(11)表示的化合物為起始物質的方式說明如下。
應予說明,所述結構式(11)表示的化合物能夠優選根據上述方法制造。
-結構式(13)表示的化合物的制造-
所述結構式(13)表示的化合物例如能夠按以下方式制造。
在所述結構式(11)表示的化合物與甲硫醇鈉的混合物中加入乙腈,在室溫下攪拌20分鐘。接著,在室溫下加入碘甲烷,進一步攪拌30分鐘。加入飽和碳酸氫鈉水溶液,用乙酸乙酯萃取。能夠對有機層進行芒硝干燥后,采用硅膠色層分離法(己烷:乙酸乙酯=2:1)純化得到的殘渣,從而獲得結構式(13)表示的化合物。
對于所述各結構式表示的化合物的制造方法中的反應條件、使用的化合物及其用量、溶劑等,只要不損害本發明的效果,就沒有特別限制,能夠根據目的適當選擇。
所述各化合物是否具有所述各結構式表示的結構能夠通過適當選擇的各種分析方法來確認,例如可以舉出所述質譜分析法、所述紫外分光法、所述紅外分光法、所述質子核磁共振分光法、所述碳13核磁共振分光法等分析方法等。
(含化合物的組合物、抗癌劑、抗幽門螺桿菌劑)
<含化合物的組合物>
本發明的含化合物的組合物至少含有所述結構式(1)~(13)中的任一個表示的化合物,根據需要地進一步含有其它成分。
所述結構式(1)~(13)中的任一個表示的化合物可以單獨使用一種,也可以將兩種以上并用。
作為所述含化合物的組合物中的以所述結構式(1)~(13)中的任一個表示的化合物的含量,沒有特別限制,能夠根據目的適當選擇。所述含化合物的組合物可以是以所述結構式(1)~(13)中的任一個表示的化合物其自身。
-其它成分-
作為所述其它成分,沒有特別限制,能夠從可在藥理學上容許的載體之中根據目的適當選擇,例如可以舉出添加劑、輔劑、水等。這些可以單獨使用一種,也可以將兩種以上并用。
作為所述添加劑或所述輔劑,沒有特別限制,能夠根據目的適當選擇,例如可以舉出殺菌劑、保鮮劑、粘結劑、增稠劑、固定劑、粘合劑、著色劑、穩定化劑、ph調節劑、緩沖劑、等滲化劑、溶劑、抗氧化劑、防紫外線劑、防晶析劑、消泡劑、物性提高劑、防腐劑等。
作為所述殺菌劑,沒有特別限制,能夠根據目的適當選擇,例如可以舉出氯化芐烷銨、苯索氯銨、十六烷基氯化吡啶鎓等陽離子性表面活性劑等。
作為所述保鮮劑,沒有特別限制,能夠根據目的適當選擇、例如可以舉出對羥基苯甲酸酯類、氯代丁醇、甲酚等。
作為所述粘結劑、增稠劑、固定劑,沒有特別限制,能夠根據目的適當選擇,例如可以舉出、淀粉、糊精、纖維素、甲基纖維素、乙基纖維素、羧甲基纖維素、羥基乙基纖維素、羥基丙基纖維素、羥基丙基甲基纖維素、羧甲基淀粉、支鏈淀粉、海藻酸鈉、海藻酸銨、海藻酸丙二醇酯、瓜耳膠、刺槐豆膠、阿拉伯樹膠、黃原酸膠、凝膠、酪蛋白、聚乙烯醇、聚環氧乙烷、聚乙二醇、乙烯·丙烯嵌段聚合物、聚丙烯酸鈉、聚乙烯吡咯烷酮等。
作為所述粘合劑,沒有特別限制,能夠根據目的適當選擇,例如可以舉出水、乙醇、丙醇、單糖漿、葡萄糖液、淀粉液、凝膠液、羧甲基纖維素、羥基丙基纖維素、羥基丙基淀粉、甲基纖維素、乙基纖維素、紫膠、磷酸鈣、聚乙烯吡咯烷酮等。
作為所述著色劑,沒有特別限制,能夠根據目的適當選擇,例如可以舉出氧化鈦、氧化鐵等。
作為所述穩定化劑,沒有特別限制,能夠根據目的適當選擇,例如可以舉出黃蓍、阿拉伯樹膠、凝膠、焦亞硫酸鈉、乙二胺四乙酸(edta)、巰基乙酸、硫羥乳酸等。
作為所述ph調節劑或所述緩沖劑,沒有特別限制,能夠根據目的適當選擇,例如可以舉出檸檬酸鈉、乙酸鈉、磷酸鈉等。
作為所述等滲化劑,沒有特別限制,能夠根據目的適當選擇,例如可以舉出氯化鈉、葡萄糖等。
作為所述含化合物的組合物中的所述其它成分的含量,沒有特別限制,能夠在不損害所述結構式(1)~(13)中的任一個表示的化合物的效果的范圍內,根據目的適當選擇。
-用途-
所述含化合物的組合物含有所述結構式(1)~(13)中的任一個表示的化合物,因此,具有優異的抗癌作用、優異的抗幽門螺桿菌活性,安全性高,例如可優選用于藥品組合物、抗癌劑、抗幽門螺桿菌劑等。
應予說明,所述含化合物的組合物可以單獨使用,也可以與以其它成分為有效成分的藥品合并使用。另外,所述含化合物的組合物可以以配合在以其它成分為有效成分的藥品中的狀態使用。
<抗癌劑>
本發明的抗癌劑至少含有所述結構式(1)~(13)中的任一個表示的化合物,根據需要地進一步含有其它成分。
所述結構式(1)~(13)中的任一個表示的化合物可以單獨使用一種,也可以將兩種以上并用。
作為所述抗癌劑中的所述結構式(1)~(13)中的任一個表示的化合物的含量,沒有特別限制,能夠根據目的適當選擇。所述抗癌劑可以為所述結構式(1)~(13)中的任一個表示的化合物其自身。
-其它成分-
作為所述其它成分,沒有特別限制,能夠根據目的適當選擇,例如可以舉出與在所述含化合物的組合物中記載的其它成分相同的成分。這些可以單獨使用一種,也可以將兩種以上并用。
作為所述抗癌劑中的所述其它成分的含量,沒有特別限制,能夠在不損害所述結構式(1)~(13)中的任一個表示的化合物的效果的范圍內,根據目的適當選擇。
-用途-
所述抗癌劑含有所述結構式(1)~(13)中的任一個表示的化合物,因此,具有優異的抗癌作用,安全性高,可優選用作胃癌、前列腺癌、肺癌、大腸癌、胰腺癌、乳腺癌等廣泛的癌的預防劑或治療劑。這些之中,可特別優選用于胃癌、大腸癌。
應予說明,所述抗癌劑可以單獨使用,也可以與以其它成分為有效成分的藥品合并使用。另外,所述抗癌劑可以以配合在以其它成分為有效成分的藥品中的狀態使用。
另外,如后述的試驗例所示,本發明的所述結構式(1)~(13)表示的化合物能夠在正常間質細胞的存在下更加抑制癌細胞的增殖。
所述抗癌劑含有所述結構式(1)~(13)中的任一個表示的化合物,因此,通過對個體給藥,能夠預防個體的癌生成或者治療患癌個體。所以,本發明也涉及以對個體給予所述抗癌劑為特征的癌的預防或治療方法。
<抗幽門螺桿菌劑>
本發明的抗幽門螺桿菌劑至少含有所述結構式(1)~(13)中的任一個表示的化合物,根據需要進一步含有其它成分。
所述結構式(1)~(13)中的任一個表示的化合物可以單獨使用一種,也可以將兩種以上并用。
作為所述抗幽門螺桿菌劑中的所述結構式(1)~(13)中的任一個表示的化合物的含量,沒有特別限制,能夠根據目的適當選擇。所述抗幽門螺桿菌劑可以為所述結構式(1)~(13)中的任一個表示的化合物其自身。
-其它成分-
作為所述其它成分,沒有特別限制,能夠根據目的適當選擇,例如可以舉出與在所述含化合物的組合物中記載的其它成分相同的成分。這些可以單獨使用一種,也可以將兩種以上并用。
作為所述抗幽門螺桿菌劑中的所述其它成分的含量,沒有特別限制,能夠在不損害所述結構式(1)~(13)中的任一個表示的化合物的效果的范圍內,根據目的適當選擇。
-用途-
所述抗幽門螺桿菌劑含有所述結構式(1)~(13)中的任一個表示的化合物,因此,具有優異的抗幽門螺桿菌活性,安全性高,可以優選用作胃潰瘍、十二指腸潰瘍等由幽門螺桿菌引起的胃和十二指腸損害的預防劑或治療劑。
應予說明,所述抗幽門螺桿菌劑可以單獨使用,也可以與以其它成分為有效成分的藥品合并使用。另外,所述抗幽門螺桿菌劑可以以配合在以其它成分為有效成分的藥品中的狀態使用。
所述抗幽門螺桿菌劑含有以所述結構式(1)~(13)中的任一個表示的化合物,因此,通過對個體給藥,能夠預防個體感染幽門螺桿菌、或治療感染幽門螺桿菌的個體。所以,本發明也涉及以對個體給予所述抗幽門螺桿菌劑為特征的由幽門螺桿菌帶來的感染病的預防或治療方法。
另外,通過將所述抗幽門螺桿菌劑對個體給藥,能夠預防由幽門螺桿菌引起的胃和十二指腸損害的發生、或治療患有由幽門螺桿菌引起的胃和十二指腸損害的個體。所以,本發明也涉及以對個體給予所述抗幽門螺桿菌劑為特征的由幽門螺桿菌引起的胃和十二指腸損害的預防或治療方法。
<劑型>
作為所述含化合物的組合物、抗癌劑、和抗幽門螺桿菌劑的劑型,沒有特別限制,能夠根據目的適當選擇,例如可以舉出固體劑、半固體劑、液體劑等。這些劑型的所述含化合物的組合物、抗癌劑、和抗幽門螺桿菌劑能夠依據常規方法制造。
-固體劑-
作為所述固體劑,沒有特別限制,能夠根據目的適當選擇,用作內用劑的情況下,例如可以舉出片劑、咀嚼片、泡騰片、口腔崩解片、糖錠、滴劑、硬膠囊劑、軟膠囊劑、顆粒劑、粉劑、丸劑、干糖漿劑、浸劑等。
所述固體劑用作外用劑的情況下,例如可以舉出栓劑、泥敷劑、硬膏劑等。
-半固體劑-
作為所述半固體劑,沒有特別限制,能夠根據目的適當選擇,用作內用劑的情況下,例如可以舉出舐劑(煎膏劑)、咀嚼膠劑、蓬松膠劑(whippingagent)、膠凍劑等。
所述半固體劑用作外用劑的情況下,例如可以舉出軟膏劑、乳膏劑、摩絲劑、吸入劑、nazar凝膠劑等。
-液體劑-
作為所述液體劑,沒有特別限制,能夠根據目的適當選擇,用作內用劑的情況下,例如可以舉出糖漿劑、飲劑、懸浮劑、醑劑等。
所述液體劑用作外用劑的情況下,例如可以舉出液劑、滴眼劑、氣霧劑、噴霧劑等。
<給藥>
作為所述含化合物的組合物、抗癌劑、和抗幽門螺桿菌劑的給藥方法、給藥量、給藥時間、和給藥對象,沒有特別限制,能夠根據目的適當選擇。
作為所述給藥方法,例如可以舉出局部給藥法、腸道給藥法、非口服給藥法等。
作為所述給藥量,沒有特別限制,能夠考慮給藥對象個體的年齡、體重、體質、癥狀、是否給予以其它成分為有效成分的藥品、藥劑等各種各樣的要因來適當選擇。
作為成為所述給藥對象的動物種類,沒有特別限制,能夠根據目的適當選擇、例如可以舉出人、猴、豬、牛、綿羊、山羊、狗、貓、小鼠、大鼠、鳥等,這些之中優選用于人。
實施例
下面,列舉本發明的制造例、試驗例對本發明具體地進行說明,但本發明不受這些制造例、試驗例任何限定。應予說明,下述試驗例的數據是獲得了相同的結果、獨立進行了兩次或三次的實驗的代表性數據。統計分析采用學生t檢驗(student'st-test)。
(制造例1)
<結構式(3)表示的化合物的制造>
按以下方式,通過化學合成制造所述結構式(3)表示的化合物。
-結構式(14)表示的化合物的制造-
氬氣氛下使氨基芐腈(15.0g,128mmol)溶解在無水四氫呋喃(以下有時稱為“thf”)300ml中,冰浴下滴加溴化乙基鎂(127ml,383mmol)。在室溫下使其攪拌12小時后,在冰浴下滴加10%鹽酸水溶液(100ml)。滴加完成后,在冰浴下加入氫氧化鈉,將ph調到7。分離有機層,用二乙基醚萃取水層。合并有機層進行芒硝干燥,蒸餾除去溶劑。將得到的殘渣采用硅膠色層分離法(己烷:乙酸乙酯=6:1)純化,獲得結構式(14)表示的化合物(9.7g,51%)。
--理化性質--
作為所述結構式(14)表示的化合物的理化性質,如下。
(1)外觀:黃色粉末
(2)熔點:42℃~43℃
(3)分子式:c9h11on
(4)高分辨質譜分析(hresi-ms)(m/z):
實驗值150.0911(m+h)+
計算值150.0913(以c9h12on計)
(5)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:3434,3331,1644,1620
(6)質子核磁共振光譜(400mhz,cdcl3):
δ=1.21(3h,q,j=7.3),2.98(2h,q,j=7.3),6.27(2h,brs),6.62-6.67(2h,m),7.25(1h,ddd,j=7.3,5.0,1.4),7.76(1h,dd,j=8.7,1.4)
(7)碳13核磁共振光譜(100mhz,cdcl3):
δ=8.70,32.27,115.70,117.30,117.84,131.10,134.05,150.21,203.33
-結構式(17)表示的化合物的制造-
氬氣氛下使所述結構式(14)表示的化合物(1當量)溶解在二氯甲烷中,加入三乙基胺(2當量)。進一步在冰浴下滴加作為酰氯的反-8-甲基-6-壬酰氯(1.1當量),在室溫下使其攪拌。用0.1n鹽酸停止反應,用二氯甲烷萃取,接著,用飽和碳酸氫鈉水溶液、食鹽水洗滌。合并有機層進行芒硝干燥,蒸餾除去溶劑。能夠采用硅膠色層分離法(己烷:乙酸乙酯)純化得到的殘渣而獲得結構式(17)表示的化合物。
--理化性質--
作為所述結構式(17)表示的化合物的理化性質,如下。
(1)外觀:無色油狀物
(2)分子式:c19h27o2n
(3)高分辨質譜分析(hresi-ms)(m/z):
實驗值324.1932(m+na)+
計算值324.1934(以c19h27o2nna計)
(4)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:3255,2956,2937,1655,969,754
(5)質子核磁共振光譜(400mhz,cdcl3):
δ=0.95(6h,d,j=6.8),1.22(3h,t,j=7.1),1.44(2h,m),1.75(2h,m),2.02(2h,m),2.22(1h,m),2.44(2h,t,j=7.3),3.07(2h,q,j=7.1),5.37(2h,m),7.09(1h,ddd,j=8.0,7.3,1.1),7.53(1h,ddd,j=8.5,7.3,1.4),7.92(1h,dd,j=8.0,1.4),8.77(1h,dd,j=8.5,1.1),11.77(1h,brs)
(6)碳13核磁共振光譜(100mhz,cdcl3):
δ=8.45,22.64,25.03,29.17,30.96,32.21,33.14,38.66,120.84,121.38,122.14,126.50,130.65,134.84,138.03,141.06,172.68,205.36
-結構式(3)表示的化合物的制造-
在所述結構式(17)表示的化合物(1當量)的1,4-二噁烷0.1m溶液中加入氫氧化鈉(3.0當量),在110℃下使其攪拌1小時~2小時。將反應溶液恢復到室溫,加入水,進一步加入1n鹽酸直至ph變成7。進一步加入己烷施加超聲波,則固體析出。抽濾固體,依次用水、己烷、接著用己烷/乙酸乙酯=1∶1的混合溶劑洗滌。能夠洗滌后使其干燥,從而獲得結構式(3)表示的化合物。
--理化性質--
作為所述結構式(3)表示的化合物的理化性質,如下。
(1)外觀:白色粉狀
(2)熔點:178℃~181℃
(3)分子式:c19h25on
(4)高分辨質譜分析(hresi-ms)(m/z):
實驗值284.2011(m+h)+
計算值284.2009(以c19h26on計)
(5)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:3064,2957,2933,1670,1638,1614,1555,1500,1371,1358,1152,1028,998,967,756,691
(6)質子核磁共振光譜(600mhz,cdcl3):
δ=0.95(6h,d,j=6.5),1.44(2h,m),1.69(2h,m),2.01(2h,q,j=6.8),2.15(3h,s),2.21(1h,m),2.70(2h,m),5.27-5.32(1h,m),5.36-5.39(1h,m),7.28(1h,ddd,j=8.2,5.8,1.0),7.32(1h,brd,j=8.2),7.52(1h,ddd,j=8.2,5.5,1.4),8.36(1h,dd,j=8.2,1.4),8.65(1h,br)
(7)碳13核磁共振光譜(150mhz,cdcl3):
δ=10.65,22.64,27.77,29.22,30.98,32.09,32.96,115.72,116.69,123.00,123.66,126.14,126.30,131.25,138.49,138.78,148.54,178.16
(制造例2)
<結構式(4)表示的化合物的制造>
按以下方式,通過化學合成制造所述結構式(4)表示的化合物。
-結構式(18)表示的化合物的制造-
氬氣氛下使所述結構式(14)表示的化合物(1當量)溶解在二氯甲烷中,加入三乙基胺(2當量)。進一步在冰浴下滴加作為酰氯的異戊酰氯(1.1當量),在室溫下使其攪拌。用0.1n鹽酸停止反應,用二氯甲烷萃取,接著,用飽和碳酸氫鈉水溶液、食鹽水洗滌。合并有機層進行芒硝干燥,蒸餾除去溶劑。能夠采用硅膠色層分離法(己烷∶乙酸乙酯)純化得到的殘渣而獲得結構式(18)表示的化合物。
--理化性質--
作為所述結構式(18)表示的化合物的理化性質,如下。
(1)外觀:無色油狀物
(2)分子式:c14h19o2n
(3)高分辨質譜分析(hresi-ms)(m/z):
實驗值256.1306(m+na)+
計算值256.1308(以c14h19o2nna計)
(4)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:3254,2959,1697,1376,754
(5)質子核磁共振光譜(400mhz,cdcl3):
δ=1.01(6h,d,j=6.4),1.22(3h,t,j=7.1),2.24(1h,m),2.31(2h,d,j=6.4),3.07(2h,q,j=7.1),7.10(1h,ddd,j=8.4,6.8,1.1),7.53(1h,ddd,j=8.5,6.8,1.4),7.93(1h,dd,j=8.0,1.4),8.78(1h,d,j=8.5),11.75(1h,brs)
(6)碳13核磁共振光譜(100mhz,cdcl3):
δ=8.45,22.46,26.20,33.15,48.09,120.81,121.38,122.16,130.59,134.85,141.02,172.14,205.40
-結構式(4)表示的化合物的制造-
在所述結構式(18)表示的化合物(1當量)的1,4-二噁烷0.1m溶液中加入氫氧化鈉(3.0當量),在110℃下使其攪拌1小時~2小時。將反應溶液恢復到室溫,加入水,進一步加入1n鹽酸直至ph變成7。進一步加入己烷施加超聲波,則固體析出。抽濾固體,依次用水、己烷、接著用己烷/乙酸乙酯=1∶1的混合溶劑洗滌。能夠洗滌后使其干燥,從而獲得結構式(4)表示的化合物。
--理化性質--
作為所述結構式(4)表示的化合物的理化性質,如下。
(1)外觀:白色粉狀
(2)熔點:240℃~244℃;
(3)分子式:c14h17on
(4)高分辨質譜分析(hresi-ms)(m/z):
實驗值216.1385(m+h)+
計算值216.1383(以c14h18on計)
(5)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:3059,2956,1636,1609,1554,1505,1369,1359,1189,998,762,695
(6)質子核磁共振光譜(400mhz,dmso-d6):
δ=0.89(6h,d,j=6.6),1.94(3h,s),1.99(1h,m),2.53(2h,d,j=7.5),7.18(1h,ddd,j=8.2,6.6,1.4),7.46(1h,d,j=8.2),7.52(1h,ddd,j=8.2,6.6,1.4),8.01(1h,dd,j=8.2,1.1),11.23(1h,brs)
(7)碳13核磁共振光譜(100mhz,dmso-d6):
δ=11.03,22.28,28.30,114.68,117.74,122.39,123.00,125.18,131.08,139.33,148.76,176.43
(制造例3)
<結構式(8)表示的化合物的制造>
按以下方式,通過化學合成制造所述結構式(8)表示的化合物。
-結構式(19)表示的化合物的制造-
氬氣氛下使所述結構式(14)表示的化合物(1當量)溶解在二氯甲烷中,加入三乙基胺(2當量)。進一步在冰浴下滴加作為酰氯的肉桂酰氯(1.1當量),在室溫下使其攪拌。用0.1n鹽酸停止反應,用二氯甲烷萃取,接著,用飽和碳酸氫鈉水溶液、食鹽水洗滌。合并有機層進行芒硝干燥,蒸餾除去溶劑。能夠采用硅膠色層分離法(己烷:乙酸乙酯)純化得到的殘渣而獲得結構式(19)表示的化合物。
--理化性質--
作為所述結構式(19)表示的化合物的理化性質,如下。
(1)外觀:白色粉狀
(2)熔點:109℃~111℃
(3)分子式:c18h17o2n
(4)高分辨質譜分析(hresi-ms)(m/z):
實驗值302.1151(m+na)+
計算值302.1152(以c18h17o2nna計)
(5)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:3223,3023,1677,1653,751,727
(6)質子核磁共振光譜(400mhz,cdcl3):
δ=1.25(3h,t,j=7.1),3.11(2h,q,j=7.1),6.65(1h,d,j=15.5),7.13(1h,ddd,j=8.0,7.1,1.1),7.36-7.44(3h,m),7.56-7.62(3h,m),7.76(1h,d,j=15.5),7.96(1h,dd,j=8.0,1.4),8.91(1h,dd,j=8.5,1.4),12.10(1h,brs)
(7)碳13核磁共振光譜(100mhz,cdcl3):
δ=8.43,33.15,121.06,121.47,122.09,122.41,128.07,128.83,129.95,130.69,134.66,134.93,141.25,142.25,164.85,205.56
-結構式(8)表示的化合物的制造-
在所述結構式(19)表示的化合物(1當量)的1,4-二噁烷0.1m溶液中加入氫氧化鈉(3.0當量),在110℃下使其攪拌1小時~2小時。將反應溶液恢復到室溫,加入水,進一步加入1n鹽酸直至ph變成7。進一步加入己烷施加超聲波,則固體析出。抽濾固體,依次用水、己烷洗滌。能夠洗滌后使其干燥,從而獲得結構式(8)表示的化合物。
--理化性質--
作為所述結構式(8)表示的化合物的理化性質,如下。
(1)外觀:黃色粉狀
(2)熔點:>260℃
(3)分子式:c18h15on
(4)高分辨質譜分析(hresi-ms)(m/z):
實驗值284.1046(m+na)+
計算值284.1046(以c18h15onna計)
(5)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:3064,2938,1628,1570,1507,1387,1359,1187,965,755,690
(6)質子核磁共振光譜(400mhz,dmso-d6):
δ=2.13(3h,s),7.21(1h,ddd,j=8.5,6.8,1.1),7.32-7.50(5h,m),7.55-7.59(1h,m),7.67-7.72(3h,m),8.03(1h,dd,j=8.2,1.4),11.20(1h,s)
(7)碳13核磁共振光譜(100mhz,dmso-d6):
δ=10.66,115.68,118.18,121.14,122.55,123.13,125.14,127.57,129.16,129.30,131.61,135.11,135.94,139.75,143.14,176.76
(制造例4)
<結構式(5)表示的化合物的制造>
按以下方式,通過化學合成制造所述結構式(5)表示的化合物。
-結構式(16)表示的化合物的制造-
氬氣氛下使所述結構式(14)表示的化合物(1當量)溶解在二氯甲烷中,加入三乙基胺(2當量)。進一步在冰浴下滴加作為酰氯的壬酰氯(1.1當量),在室溫下使其攪拌。用0.1n鹽酸停止反應、用二氯甲烷萃取,接著,用飽和碳酸氫鈉水溶液、食鹽水洗滌。合并有機層進行芒硝干燥,蒸餾除去溶劑。能夠采用硅膠色層分離法(己烷:乙酸乙酯)純化得到的殘渣而獲得結構式(16)表示的化合物。
--理化性質--
作為所述結構式(16)表示的化合物的理化性質,如下。
(1)外觀:無色油狀物
(2)分子式:c18h27o2n
(3)高分辨質譜分析(hresi-ms)(m/z):
實驗值312.1933(m+na)+
計算值312.1934(以c18h27o2nna計)
(4)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:3255,2953,2928,1698.754
(5)質子核磁共振光譜(400mhz,cdcl3):
δ=0.87(3h,t,j=6.6),1.20-1.41(13h,m),1.74(2h,m),2.43(2h,t,j=7.6),3.08(2h,q,j=7.3),7.09(1h,ddd,j=8.0,7.3,1.1),7.53(1h,ddd,j=8.5,7.3,1.6),7.93(1h,dd,j=8.0,1.6),8.77(1h,dd,j=8.5,1.1),11.76(1h,brs)
(6)碳13核磁共振光譜(100mhz,cdcl3):
δ=8.54,14.07,22.63,25.55,29.14,29.22,29.28,31.87,33.20,38.80,120.85,121.96,122.12,130.57,134.84,141.08,172.80,205.37
-結構式(21)表示的化合物的制造-
在所述結構式(16)表示的化合物(1當量)的1,4-二噁烷0.1m溶液中加入氫氧化鈉(3.0當量),在110℃下使其攪拌1小時~2小時。將反應溶液恢復到室溫,加入水,進一步加入1n鹽酸直至ph變成7。進一步加入己烷施加超聲波,則固體析出。抽濾固體,用水、己烷洗滌。能夠洗滌后使其干燥,從而獲得結構式(21)表示的化合物。
--理化性質--
作為所述結構式(21)表示的化合物的理化性質,如下。
(1)外觀:白色粉狀
(2)熔點:228℃~231℃
(3)分子式:c18h25on
(4)高分辨質譜分析(hresi-ms)(m/z):
實驗值272.2010(m+h)+
計算值272.2009(以c18h26on計)
(5)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:3059,2952,2923,1638,1607,1555,1500,1190,998,754,693
(6)質子核磁共振光譜(400mhz,dmso-d6):
δ=0.80(3h,t,j=6.6),1.19-1.35(10h,m),1.59(2h,m),1.94(3h,s),2.62(2h,t,j=7.5),7.18(1h,dd,j=8.2,6.6),7.45(1h,d,j=8.0),7.51(1h,ddd,j=8.2,6.7,1.4),8.00(1h,dd,j=8.0,1.4),11.31(1h,brs)
(7)碳13核磁共振光譜(100mhz,dmso-d6):
δ=10.48,14.15,22.26,28.49,28.80,28.92,29.04,31.43,31.84,113.87,117.73,122.39,123.06,125.18,131.05,139.36,149.82,176.39
-結構式(5)表示的化合物的制造-
氬氣氛下使所述結構式(21)表示的化合物(800mg,2.95mmol)溶解在thf(20ml)中,加入叔丁醇鋰的thf溶液(4.4ml,4.42mmol),在室溫下使其攪拌20分鐘。接著,加入溴乙酸甲酯(1.4ml、14.7mmol),進一步使其回流下攪拌12小時。加入水,使反應停止,用乙酸乙酯萃取。對有機層進行芒硝干燥,蒸餾除去溶劑。將得到的殘渣采用硅膠色層分離法(己烷:乙酸乙酯=2∶1)純化,從而獲得結構式(5)表示的化合物(780mg,76%)。
--理化性質--
作為所述結構式(5)表示的化合物的理化性質,如下。
(1)外觀:無色油狀物
(2)分子式:c21h29o3n
(3)高分辨質譜分析(hresi-ms)(m/z):
實驗值344.2222(m+h)+
計算值344.2220(以c21h30o3n計)
(4)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:2953,2922,1743,1635,1617,1558,1507,1214,994,760,688
(5)質子核磁共振光譜(400mhz,cdcl3):
δ=0.89(3h,t,j=6.6),1.21-1.51(10h,m),1.60(2h,m),2.22(3h,s),2.74(2h,br),3.80(3h,s),4.90(2h,s),7.20(1h,d,j=8.7),7.33(1h,ddd,j=8.0,6.8,0.7),7.58(1h,ddd,j=8.7,7.1,1.6),8.47(1h,dd,j=8.0,1.6)
(6)碳13核磁共振光譜(100mhz,cdcl3):
δ=11.65,14.06,22.60,28.06,29.13,29.17,29.75,30.93,31.75,48.47,53.01,114.19,117.55,123.11,124.79,127.30,131.89,140.66,150.66,168.69,177.39
(制造例5)
<結構式(6)表示的化合物的制造>
按以下方式,通過化學合成制造所述結構式(6)表示的化合物。
-結構式(6)表示的化合物的制造-
使所述結構式(5)表示的化合物(890mg,2.59mm0l)溶解在乙醇(以下有時稱為“etoh”。5ml)與thf(5ml)的混合溶劑中,加入2m氫氧化鈉水溶液(2.0ml),在室溫下使其攪拌2小時。冰浴下加入1n鹽酸將ph調到4,用乙酸乙酯萃取。對有機層進行芒硝干燥,使溶劑蒸餾除去,從而獲得結構式(6)表示的化合物(530mg,62%)。
--理化性質--
作為所述結構式(6)表示的化合物的理化性質,如下。
(1)外觀:白色粉狀
(2)熔點:161℃~163℃
(3)分子式:c20h27o3n
(4)高分辨質譜分析(hresi-ms)(m/z):
實驗值330.2063(m+h)+
計算值330.2064(以c20h28o3n計)
(5)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:2955,2925,2853,1725,1635,1593,1506,1191,976,760,689
(6)質子核磁共振光譜(400mhz,cdcl3):
δ=0.87(3h,t,j=6.4),1.20-1.65(12h,m),2.19(3h,s),2.80(2h,br),4.98(2h,br),7.26(1h,t,j=8.0),7.42(1h,d,j=8.7),7.54(1h,ddd,j=8.7,6.8,1.1),8.39(1h,dd,j=8.0,1.1)
(7)碳13核磁共振光譜(100mhz,cdcl3):
δ=11.91,14.06,22.68,27.85,29.12,29.78,31.26,31.73,49.71,115.46,117.21,123.86,123.94,126.69,132.47,140.53,154.22,169.35.176.57
(制造例6)
<結構式(1)表示的化合物的制造>
按以下方式,通過化學合成制造所述結構式(1)表示的化合物。
-結構式(1)表示的化合物的制造-
使所述結構式(21)(100mg,0.37mmol)溶解在n,n-二甲基甲酰胺(以下有時稱為“dmf”。5ml),加入碳酸鉀(332mg,2.40mmol)、溴乙酸甲酯(53.0ml,0.56mmol),在80℃下使其攪拌12小時。加入水,使反應停止,用乙酸乙酯萃取。對有機層進行芒硝干燥,蒸餾除去溶劑。將得到的殘渣采用硅膠色層分離法(己烷:乙酸乙酯=2∶1)純化,獲得結構式(1)表示的化合物(83.0mg,66%)。
--理化性質--
作為所述結構式(1)表示的化合物的理化性質,如下。
(1)外觀:無色油狀物
(2)分子式:c21h29o3n
(3)高分辨質譜分析(hresi-ms)(m/z):
實驗值344.2221(m+h)+
計算值344.2220(以c21h30o3n計)
(4)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:2953,2925,2854,1765,1618,1596,1437,1123,968,768,680
(5)質子核磁共振光譜(400mhz,cdcl3):
δ=0.87(3h,t,j=6.6),1.20-1.48(10h,m),1.71(2h,m),2.43(3h,s),2.95(2h,m),3.86(3h,s),4.62(2h,s),7.47(1h,ddd,j=8.2,6.8,1.1),7.62(1h,ddd,j=8.4,6.8,1.3),8.00(1h,d,j=8.4),8.05(1h,d,j=8.2)
(6)碳13核磁共振光譜(100mhz,cdcl3):
δ=11.90,14.08,22.63,28.99,29.23,29.49,29.86,31.83,37.02,52.32,70.26,120.85,120.73,121.39,121.76,125.73,128.82,128.85,147.84,159.03,164.32,168.95
(制造例7)
<結構式(2)表示的化合物的制造>
按以下方式,通過化學合成制造所述結構式(2)表示的化合物。
-結構式(2)表示的化合物的制造-
使所述結構式(1)表示的化合物(80.0mg,0.233mmol)溶解在etoh(1ml)與thf(1ml)的混合溶劑中,加入2m氫氧化鈉水溶液(0.5ml),在室溫下使其攪拌2小時。冰浴下加入1n鹽酸將ph調成4,用乙酸乙酯萃取。對有機層進行芒硝干燥,使溶劑蒸餾除去,獲得結構式(2)表示的化合物(68.2mg,88%)。
--理化性質--
作為所述結構式(2)表示的化合物的理化性質,如下。
(1)外觀:白色粉狀
(2)熔點:59℃~62℃
(3)分子式:c20h27o3n
(4)高分辨質譜分析(hresi-ms)(m/z):
實驗值330.2064(m+h)+
計算值330.2064(以c20h28o3n計)
(5)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:2927,2855,2713,1736,1642,1589,1227,1181,1078,764,724
(6)質子核磁共振光譜(600mhz,methanol-d4):
δ=0.88(3h,t,j=6.8),1.25-1.41(8h,m),1.50(2h,m),1.78(2h,m),2.54(3h,s),3.15(2h,t,m),4.92(2h,s),7.54(1h,ddd,j=8.2,7.2,1.0),7.92(1h,ddd,j=8.5,6.8,1.0),8.07(1h,brd,j=8.5),8.42(1h,brd,j=8.2)
(7)碳13核磁共振光譜(150mhz,methanol-d4):
δ=12.23,14.40,23.67,29.93,30.27,30.34,30.74,32.97,35.07,72.72,122.81,123.71,123.85,124.62,129.16,133.85,142.14,164.30,167.46,171.91
(制造例8)
<結構式(7)表示的化合物的制造>
按以下方式,通過化學合成制造所述結構式(7)表示的化合物。
-結構式(7)表示的化合物的制造-
使所述結構式(21)表示的化合物(50.0mg,0.184mmol)溶解在thf(1.0ml)中,加入叔丁醇鋰(29.4mg,0.37mmol),在室溫下使其攪拌20分鐘。接著,加入溴化氰(0.6ml,1.84mmol),在室溫下使其攪拌2小時。加入水,使反應停止,用乙酸乙酯萃取。對有機層進行芒硝干燥,蒸餾除去溶劑。將得到的殘渣采用硅膠色層分離法(己烷:乙酸乙酯=2:1)純化,獲得結構式(7)表示的化合物(34.0mg,62%)。
--理化性質--
作為所述結構式(7)表示的化合物的理化性質,如下。
(1)外觀:無色油狀物
(2)分子式:c19h24on2
(3)高分辨質譜分析(hresi-ms)(m/z):
實驗值297.1961(m+h)+
計算值297.1961(以c19h25on2計)
(4)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:2961,2926,28532237,1628,1576,1470,1292,1191,761,693
(5)質子核磁共振光譜(400mhz,cdcl3):
δ=0.87(3h,t,j=6.8),1.20-1.50(10h,m),1.71(2h,m),2.15(3h,s),2.93(2h,m),7.47(1h,m),7.73(2h,m),8.33(1h,ddd,j=8.0,0.92,1.1)
(6)碳13核磁共振光譜(100mhz,cdcl3):
δ=11.20,14.05,22.59,28.0o,29.07,29.11,29.42,31.73,31.80,106.42,116.28,120.30,123.35,126.23,127.08,133.34,137.25,146.10.177.31
(制造例9)
<結構式(9)表示的化合物的制造>
按以下方式,通過化學合成制造所述結構式(9)表示的化合物。
-結構式(9)表示的化合物的制造-
使所述結構式(6)表示的化合物(100mg,0.30mmol)懸浮在thf(5ml)中,加入三乙基胺(50.8ml,0.36mmol)、疊氮磷酸二苯酯(以下有時稱為“dppa”。72.0ml,0.33mmol)、甲硫醇鈉(23.0mg,0.334mmol),回流下攪拌2小時。加入氯化銨水溶液,用乙酸乙酯萃取。對有機層進行芒硝干燥,蒸餾除去溶劑。將得到的殘渣采用硅膠色層分離法(己烷:乙酸乙酯=3∶1)純化,獲得結構式(9)表示的化合物(39.3mg,36%)。
--理化性質--
作為所述結構式(9)表示的化合物的理化性質,如下。
(1)外觀:無色油狀物
(2)分子式:c21h29o2ns
(3)高分辨質譜分析(hresi-ms)(m/z):
實驗值360.1994(m+h)+
計算值360.1992(以c21h30o2ns計)
(4)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:2924,2852,1687,1614,1594,1542,1193,1028,757.558
(5)質子核磁共振光譜(400mhz,cdcl3):
δ=0.88(3h,t,j=6.4),1.20-1.50(10h,m),1.60(2h,br),2.23(3h,s),2.33(3h,s),2.51-2.99(2h,br),5.01(2h,br),7.21(1h,d,j=8.7),7.34(1h,dd,j=8.0、6.6),7.58(1h,ddd,j=8.7,6.6,1.4),8.47(1h,dd,j=8.0,1.4)
(6)碳13核磁共振光譜(100mhz,cdcl3):
δ=11.38,11.68,14.06,22.60,28.16,29.13,29.16,29.75,31.03,31.74,55.95,114.61,117.94,123.32,127.29,131.97,132.02,140.73,150.65,177.49,196.82
(制造例10)
<結構式(10)表示的化合物的制造>
按以下方式,通過化學合成制造所述結構式(10)表示的化合物。
-結構式(10)表示的化合物的制造-
使所述結構式(6)表示的化合物(100mg,0.30mmol)懸浮在thf(5ml)中,在冰冷卻下加入三乙基胺(50.8ml,0.365mmol)、dppa(72.0ml,0.33mmol),進一步回流下攪拌1小時。在其中加入甲硫醇鈉(23.0mg,0.33mmol),進一步攪拌1小時。加入氯化銨水溶液,用乙酸乙酯萃取。對有機層進行芒硝干燥,蒸餾除去溶劑。將得到的殘渣采用硅膠色層分離法(己烷:乙酸乙酯=5∶1)純化,獲得結構式(10)表示的化合物(48.6mg,43%)。
--理化性質--
作為所述結構式(10)表示的化合物的理化性質,如下。
(1)外觀:無色油狀物
(2)分子式:c21h30o2n2s
(3)高分辨質譜分析(hresi-ms)(m/z):
實驗值397.1921(m+na)+
計算值397.1920(以c21h30o2n2nas計)
(4)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:3169,2955,2927,1671,1615,1595,1556,1492,1195,1084,760,651
(5)質子核磁共振光譜(600mhz,cdcl3):
δ=0.89(3h,t,j=6.7),1.22-1.45(15h,m),2.45(2h,br),2.46(3h,s),5.67(2h,br),7.24(1h,ddd,j=7.9,6.8,1.0),7.49(1h,d,j=8.6),7.59(1h,ddd,j=8.6,6.8,1.4),8.26(1h,dd,j=7.9,1.4),8.78(1h,br)
(6)碳13核磁共振光譜(150mhz,cdcl3):
δ=11.05,12.22,14.02,22.59,28.48,29.11,29.16,29.73,30.73,31.77,52.58,115.46,117.00,123.24,124.37,126.89,132.32,139.56,151.60,168.61,177.27
(制造例11)
<結構式(11)表示的化合物的制造>
按以下方式,通過化學合成制造所述結構式(11)表示的化合物。
-結構式(15)表示的化合物的制造-
氬氣氛下使所述結構式(14)表示的化合物(1當量)溶解在二氯甲烷中,加入三乙基胺(2當量)。進一步在冰浴下滴加作為酰氯的辛酰氯(3.0當量),在室溫下使其攪拌。用0.1n鹽酸停止反應,用二氯甲烷萃取,接著,用飽和碳酸氫鈉水溶液、食鹽水洗滌。合并有機層進行芒硝干燥,蒸餾除去溶劑。能夠采用硅膠色層分離法(己烷:乙酸乙酯)純化得到的殘渣而獲得結構式(15)表示的化合物。
--理化性質--
作為所述結構式(15)表示的化合物的理化性質,如下。
(1)外觀:無色油狀物
(2)分子式:c17h25o2n
(3)高分辨質譜分析(hresi-ms)(m/z):
實驗值298.1777(m+na)+
計算值298.1778(以c17h25o2nna計)
(4)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:3255,2954,2928,1698,754
(5)質子核磁共振光譜(400mhz,cdcl3):
δ=0.87(3h,t,j=6.6),1.20-1.41(11h,m),1.74(2h,m),2.43(2h,t,j=7.5),3.08(2h,q,j=7.1),7.09(1h,ddd,j=8.2,7.1,1.4),7.53(1h,ddd,j=8.4,7.1,1.4),7.92(1h,dd,j=8.2,1.4),8.77(1h,dd,j=8.4,1.4),11.65(1h,br)
(6)碳13核磁共振光譜(100mhz,cdcl3):
δ=8.44,14.05,22.59,25.53,28.98,29.16,31.67,33.13,38.78,120.83,121.37,122.11,130.56,134.82,141.06,172.78,205.36
-結構式(20)表示的化合物的制造-
在所述結構式(15)表示的化合物(1當量)的二噁烷0.1m溶液中加入氫氧化鈉(3.0當量),在110℃下使其攪拌1小時~2小時。將反應溶液恢復到室溫,加入水,進一步加入1n鹽酸直至ph變成7。進一步加入己烷施加超聲波,則固體析出。抽濾固體,用水、己烷洗滌。能夠洗滌后使其干燥,從而獲得結構式(20)表示的化合物。
--理化性質--
作為所述結構式(20)表示的化合物的理化性質,如下。
(1)外觀:白色粉狀
(2)熔點:228℃~231℃
(3)分子式:c17h23on
(4)高分辨質譜分析(hresi-ms)(m/z):
實驗值258.1853(m+h)+
計算值258.1852(以c17h24on計)
(5)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:3061,2954,2925,1637,1608,1556,1504,1188,998,754,692
(6)質子核磁共振光譜(400mhz,methanol-d4):
δ=0.88(3h,t,j=6.6),1.30一1.46(8h,m),1.65-1.73(2h,m),2.14(3h,s),2.78(2h,t,j=7.7),7.32(1h,t,j=8.1),7.52(1h,d,m),7.61(1h,m),8.21(1h,d,j=8.2)
(7)碳13核磁共振光譜(100mhz,methanol-d4):
δ=10.84,14.40,23.68,29.99,30.16,30.54,32.91,33.45,116.16,118.67,124.38,124.51,126.15,132.65,140.55,153.37,179.53
-結構式(11)表示的化合物的制造-
氬氣氛下使所述結構式(20)表示的化合物(1.0g,3.89mmol)溶解在thf(10ml)中,加入叔丁醇鋰的thf溶液(5.8ml,5.80mmol),在室溫下使其攪拌20分鐘。冰冷卻下在其中滴加硫氰酸氯甲酯(6.7ml,38.9mmol),進一步在室溫下使其攪拌2小時。加入食鹽水,使反應停止,用乙酸乙酯萃取。對有機層進行芒硝干燥后,將得到的殘渣采用硅膠色層分離法(己烷:乙酸乙酯=2∶1)純化,獲得結構式(11)表示的化合物(330mg,26%)。
--理化性質--
作為所述結構式(11)表示的化合物的理化性質,如下。
(1)外觀:黃色油狀物
(2)分子式:c19h24on2s
(3)高分辨質譜分析(hresi-ms)(m/z):
實驗值329.1682(m+h)+
計算值329.1682(以c19h25on2s計)
(4)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:2961,2926,2853,2237,1628,1576,1470,1292,1191,987,761,693
(5)質子核磁共振光譜(400mhz,cdcl3):
δ=0.90(3h,t,j=6.6),1.23-1.71(10h,m),2.19(3h,s),2.84(2h,m),5.71(2h,s),7.38(1h,ddd,j=8.0,6.8,0.9),7.46(1h,d,j=8.7),7.68(1h,ddd,j=8.7,6.8,1.6),8.45(1h,dd,j=8.0,1.6)
(6)碳13核磁共振光譜(100mhz,cdcl3):
δ=11.60,14.05,22.56,28.59,28.88,29.76,30.55,31.66,56.26,114.42,118.19,123.83,124.60,127.31,132.41,139.86,141.68,149.60.177.64
(制造例12)
<結構式(12)表示的化合物的制造>
按以下方式,通過化學合成制造所述結構式(12)表示的化合物。
-結構式(12)表示的化合物的制造-
在所述結構式(11)表示的化合物(100mg,0.30mmol)與甲硫醇鈉(23.4mg,0.33mmol)的混合物中加入乙腈(1.5ml),在室溫下攪拌20分鐘。加入飽和碳酸氫鈉水溶液,用乙酸乙酯萃取。對有機層進行芒硝干燥后,將得到的殘渣采用硅膠色層分離法(己烷:乙酸乙酯=2∶1)純化,獲得結構式(12)表示的化合物(31.7mg,27%)。
--理化性質--
作為所述結構式(12)表示的化合物的理化性質,如下。
(1)外觀:黃色粉狀
(2)熔點:167℃~170℃;
(3)分子式:c20h28on2s2
(4)高分辨質譜分析(hresi-ms)(m/z):
實驗值399.1534(m+na)+
計算值399.1535(以c20h28on2nas2計)
(5)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:3119,2958,2918,2850,1619,1598,1538,1282,1199,1105,938,764,688
(6)質子核磁共振光譜(400mhz,cdcl3):
δ=0.86(3h,t,j=6.6),1.21-1.50(13h,m),2.22-2.58(2h,br),2.76(3h,s),5.68-6.41(2h,br),7.22(1h,t,j=7.8),7.44(1h,d,j=8.7),7.59(1h,ddd,j=8.7,7.8,1.1),8.15(1h,d,j=7.8),10.09(1h,br)
(7)碳13核磁共振光譜(100mhz,cdcl3):
δ=10.74,14.02,18.01,22.53,28.29,28.77,29.64,30.85,31.59,58.21,115.93,116.84,123.54,123.91,126.35,132.65,139.39,152.23,177.13,199.84
(制造例13)
<結構式(13)表示的化合物的制造>
按以下方式,通過化學合成制造所述結構式(13)表示的化合物。
-結構式(13)表示的化合物的制造-
在所述結構式(11)表示的化合物(50.0mg,0.15mmol)與甲硫醇鈉(10.6mg,0.15mmol)的混合物中加入乙腈(1.5ml),在室溫下攪拌20分鐘。接著,在室溫下加入碘甲烷(8.5ml,0.17mmol),進一步攪拌30分鐘。加入飽和碳酸氫鈉水溶液,用乙酸乙酯萃取。對有機層進行芒硝干燥后,將得到的殘渣采用硅膠色層分離法(己烷:乙酸乙酯=2:1)純化,獲得結構式(13)表示的化合物(30.6mg,51%)。
--理化性質--
作為所述結構式(13)表示的化合物的理化性質,如下。
(1)外觀:白色粉狀
(2)熔點:92℃~94℃
(3)分子式:c21h30on2s2
(4)高分辨質譜分析(hresi-ms)(m/z):
實驗值413.1689(m+na)+
計算值413.1692(以c21h30on2nas2計)
(5)紅外線吸收光譜:
經kbr片劑法測定的紅外線吸收的峰如下。
νmax(kbr)cm-1:2958,2922,2852,1618,1595,1566,1492,1370,1277,1192,1004,769,700
(6)質子核磁共振光譜(400mhz,cdcl3):
δ=0.89(3h,t,j=6.8),1.24-1.50(8h,m),1.64(2h,m),2.22(3h,s),2.28(3h,s),2.71(3h,s),2.77(2h,m),5.60(2h,s),7.31(2h,m),7.55(1h,ddd,j=8.4,7.1,1.6),8.46(1h,dd,j=8.0,1.6)
(7)碳13核磁共振光譜(100mhz,cdcl3):
δ=11.50,14.06,14.75,15.03,22.61,28.28,28.92,29.83,30.66,31.76,63.75,115.84,116.97,122.77,124.81,126.75,131.27,141.06,151.35,161.39,177.54
(試驗例1:體外(invitro)的抗癌活性)
將所述制造例1~13中得到的結構式(1)~(13)表示的化合物的invitro的抗癌活性按以下方式進行試驗。
<細胞增殖試驗1(人胃癌細胞mkn-74)>
-細胞的制備-
將人胃癌細胞mkn-74(理研cellbank)在添加有10%fbs(gibco公司制)、100units/ml的青霉素g(invitrogen公司制)、100μg/ml的鏈霉素(invitrogen公司制)的dmem中在37℃、5%co2下培養。
利用lipofectamine試劑(invitrogen公司制)對所述癌細胞基因轉入綠色熒光蛋白(greenfluorescenceprotein,gfp)表達載體pegfp-c1(bdbiosciences公司制),將穩定地表達gfp的細胞克隆。
將來自人胃的正常間質細胞hs738((crl-7869),atcc)在添加有10%fbs、100units/ml的青霉素g(invitrogen公司制)、100μg/ml的鏈霉素(invitrogen公司制)、5μg/ml的胰島素、5μg/ml的轉鐵蛋白(和光純藥工業公司制)、1.4μm的氫化可的松(sigma公司制)、5mg/ml的basic-fgf(peprotech公司制)的dmem中在37℃、5%co2下培養。
-共培養試驗-
使所述來自人胃的正常間質細胞hs738在含有1%透析血清、5μg/ml的胰島素、5μg/ml的轉鐵蛋白(和光純藥工業公司制)、1.4μm的氫化可的松(sigma公司制)的dmem中以5×104個/ml分散,以0.1ml/孔鋪撒在96孔板中,在各濃度下加入各評價樣品(結構式(1)~(13)表示的化合物),在37℃、5%co2下培養2天。
接著,使所述人胃癌細胞mkn-74在dmem中以5×105個/ml分散,以10μl/孔鋪撒在培養了所述來自人胃的正常間質細胞hs738的板中,進一步在37℃、5%co2下共培養3天。
-單獨培養試驗-
僅將含有1%透析血清、5μg/ml的胰島素、5μg/ml的轉鐵蛋白(和光純藥工業公司制)、1.4μm的氫化可的松(sigma公司制)的dmem以0.1ml/孔鋪撒在96孔板中,在各濃度下加入各評價樣品(結構式(1)~(13)表示的化合物),在37℃、5%co2下保持2天。
接著,使所述人胃癌細胞mkn-74在dmem中以5×105個/ml分散,以10μl/孔鋪撒在所述板中,進一步在37℃、5%co2下共培養3天。
-細胞增殖率的測定-
所述共培養試驗和單獨培養試驗中細胞增殖率的測定按以下方式進行。
從所述板的孔中除去培養基,以0.1ml/孔加入細胞裂解液(10mmtris-hcl[ph7.4]、150mmnacl、0.9mmcacl2、1%tritonx-100),裂解細胞,在激發波長為485nm、熒光波長為538nm下測定gfp的熒光強度,由下述式計算出細胞增殖率。將由所述細胞增殖率的結果計算出ic50的結果示于表1。
細胞增殖率(%)=(有評價樣品的熒光強度/沒有評價樣品的熒光強度)×100
[表1]
表1中的值表示2系列的平均值,標準誤差(se)為10%以下。
表1中,“mono”表示單獨培養試驗的結果,“cocul”表示共培養試驗的結果。
如表1所示,對于所述結構式(1)~(13)表示的13種化合物,均與僅培養胃癌細胞mkn-74的(mono)進行比較,以更低濃度的ic50值強烈地阻礙與胃間質細胞共培養時(cocul)的胃癌細胞mkn-74的增殖。
(試驗例2:體內(invivo)的抗癌活性)
將所述制造例中得到的結構式(2)表示的化合物、和結構式(13)表示的化合物的invivo的抗癌活性按以下方式試驗。
<試驗例2-1:單獨人胃癌細胞mkn-74>
將balb/cnu/nu裸鼠(雌,5周齡,charlesriver公司制)在spf條件下飼養。
對培養的人胃癌細胞mkn-74進行胰蛋白酶處理,將從培養皿中剝落的所述人胃癌細胞mkn-74(8×106個)分散在含有0.3ml的10%fbs的dmem中,與0.5ml的減少生長因子基底膜(growthfactor-reducedmatrigel,bdbiosciences公司制)混合。
將所述混合的細胞液0.1ml(癌細胞1×106個)皮下接種在所述小鼠的左鼠脛部中。
將所述結構式(2)表示的化合物、或結構式(13)表示的化合物在規定的時間向靜脈內給藥,切下皮下出現的腫瘤,測定其重量。應予說明,所述結構式(2)表示的化合物、或結構式(13)表示的化合物的給藥量以每個給藥日的量計為12.5mg/kg。
另外,腫瘤體積參照所述非專利文獻1,由以下式計算出。
腫瘤體積(mm3)=(長徑×短徑2)/2
應予說明,對給予了生理食鹽水來代替結構式(2)表示的化合物、或結構式(13)表示的化合物的(vehicle)也同樣地進行試驗作為對照。
<試驗例2-2:人胃癌細胞mkn-74和來自人胃的正常間質細胞hs738>
在試驗例2-1中,使單獨使用人胃癌細胞mkn-74這點為人胃癌細胞mkn-74和來自人胃的正常間質細胞hs738,除此之外,與試驗例2-1同樣地進行試驗。
應予說明,細胞液和對小鼠接種細胞液按以下方式進行。
分別對培養的人胃癌細胞mkn-74和來自人胃的正常間質細胞hs738進行胰蛋白酶處理,從培養皿中剝落。將所述人胃癌細胞mkn-74(8×106個)、和所述來自人胃的正常間質細胞hs738(8×106個)分散在含有0.3ml的10%fbs的dmem中,與0.5ml的減少生長因子基底膜(growthfactor-reducedmatrigel,bdbiosciences公司制)混合。
將所述混合的細胞液0.1ml(癌細胞1×106個、和間質細胞1×106個的混合)皮下接種在所述小鼠的左鼠脛部。
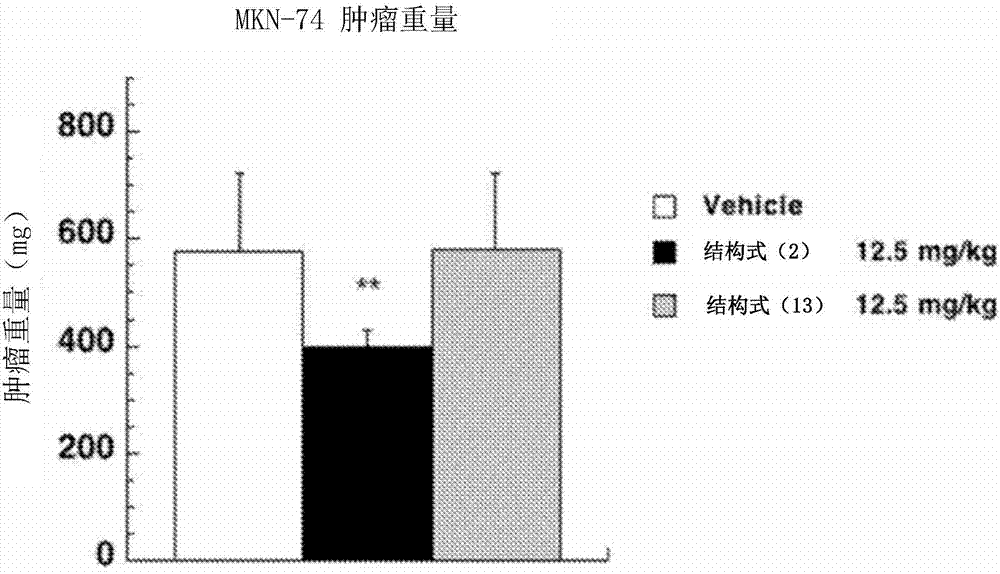
將所述試驗例2的結果示于圖1a~圖1d。
圖1a表示所述試驗例2-1中的腫瘤體積的變化,圖1b表示所述試驗例2-1中的腫瘤重量(從腫瘤接種起第21天),圖1c表示所述試驗例2-2中的腫瘤體積的變化,圖1d表示所述試驗例2-2中的腫瘤重量(從腫瘤接種起第21天)。
圖1a和圖1c中,“○”表示“vehicle”的結果,“●”表示“結構式(2)表示的化合物的給藥量為12.5mg/kg”的結果,“□”表示“結構式(13)表示的化合物的給藥量為12.5mg/kg”的結果。另外,圖1a和圖1c中,“箭頭”表示將結構式(2)表示的化合物、或結構式(13)表示的化合物給藥的天數。
圖1b和圖1d中,“白”表示“vehicle”的結果,“黑”表示“結構式(2)表示的化合物的給藥量為12.5mg/kg”的結果,“灰”表示“結構式(13)表示的化合物的給藥量為12.5mg/kg”的結果。
圖1a~圖1d中的值表示5只小鼠的平均值與標準偏差(sd),*表示p<0.05,**表示p<0.01。
如圖1a~圖1d所示,對于單獨人胃癌細胞mkn-74來說,通過將結構式(2)表示的化合物以12.5mg/kg靜脈內給藥而有意義地抑制。
另一方面,對于與來自人胃的間質細胞hs738一起移植的腫瘤來說,通過將結構式(13)表示的化合物以12.5mg/kg靜脈內給藥而有意義抑制。
(試驗例3:急性毒性試驗)
將icr小鼠(雌,4周齡,charlesriver公司制)在spf條件下飼養。
作為評價樣品,將所述結構式(1)~(13)表示的化合物進行靜脈內給藥并觀察小鼠2周。2周的觀察期間中,以確認了死亡或重度的毒性的給藥量的二分之一量作為本實驗中的最大耐容量(mtd)。將結果示于表2。
[表2]
由所述表2的結果可知,將所述結構式(1)表示的化合物、結構式(2)表示的化合物、結構式(6)表示的化合物、結構式(9)表示的化合物、結構式(10)表示的化合物、結構式(12)表示的化合物、結構式(13)表示的化合物進行靜脈內給藥的情況下的mtd為50mg/kg以上。
(試驗例4:抗菌活性)
將所述制造例中得到的結構式(2)表示的化合物、結構式(3)表示的化合物、結構式(4)表示的化合物、結構式(5)表示的化合物、結構式(6)表示的化合物、結構式(8)表示的化合物、結構式(11)表示的化合物、結構式(12)表示的化合物、和結構式(13)表示的化合物的抗菌活性按以下方式進行試驗。
應予說明,作為比較,對于克拉霉素(clarithromycin)、和氨芐青霉素(abpc)也同樣地進行試驗。
<試驗例4-1:針對幽門螺桿菌測定mic>
進行所述各化合物針對幽門螺桿菌的最小抑菌濃度(mic)的測定。
將幽門螺桿菌(helicobacterpylori)jcm12093株、和幽門螺桿菌(h.pylori)jcm12095株在hp培養基(在腦心浸液肉湯培養基(bectondickinson公司制)中添加10%胎牛血清(lifetechnologies公司制))中在37℃、微需氧培養條件下(微需氧條件(n2:o2:co2=85:5:10))靜置培養144小時。培養完成后,將培養液在hp培養基中懸浮,以幽門螺桿菌為2×106cfu/ml~9×106cfu/ml的方式稀釋。
各試驗樣品(所述各化合物、克拉霉素、氨芐青霉素)分別在hp培養基中以256mg/l制備。由此,進行兩倍階段稀釋,進行15階段的稀釋到0.0078mg/l為止。
分別在含有所述各濃度的試驗樣品的50μl/孔的hp培養基中以50μl/孔添加所述稀釋了的各菌液,在37℃、微需氧培養條件下(微需氧條件(n2:o2:co2=85:5:10))下靜置培養144小時。
培養完成后,通過濁度目視判斷各菌是否增殖,求得各菌株的mic。將結果示于表3。
<試驗例4-2:針對黃色葡萄球菌和大腸桿菌測定mic>
進行所述各化合物針對黃色葡萄球菌和大腸桿菌的最小抑菌濃度(mic)的測定。
將金黃色葡萄球菌(staphylococcusaureus)fda209p株、和大腸桿菌(escherichiacoli)k-12株在普通肉湯培養基(polypeptone(日本制藥公司制)1%、細菌用魚提取物(極東制藥工業公司制)1%、氯化鈉0.2%)中在37℃下振蕩培養一夜。培養完成后在普通肉湯培養基中以各細菌為2×106cfu/ml~9×106cfu/ml的方式稀釋。
各試驗樣品(所述各化合物、克拉霉素、氨芐青霉素)分別在普通肉湯培養基中以256mg/l制備。由此,進行兩倍階段稀釋,進行11階段的稀釋到0.125mg/l為止。
分別在含有所述各濃度的試驗樣品的50μl/孔的普通肉湯培養基中以50μl/孔添加所述稀釋了的各菌液,在37℃下靜置培養一夜。
培養完成后,通過濁度目視判斷各菌是否增殖,求得各菌株的mic。將結果示于表3。
<試驗例4-3:針對腸球菌測定mic>
進行所述各化合物針對腸球菌的最小抑菌濃度(mic)的測定。
將糞腸球菌(enterococcusfaecalis)jcm5803株在心浸湯培養基(bectondickinson公司制)中在37℃下振蕩培養一夜。培養完成后,用心浸湯培養基稀釋,以各細菌為2×104cfu/ml~9×104cfu/ml的方式稀釋。
各試驗樣品(所述各化合物、克拉霉素、氨芐青霉素)分別在心浸湯培養基中以256mg/l制備。由此,進行兩倍階段稀釋,進行11階段的稀釋到0.125mg/l為止。
分別在含有所述各濃度的試驗樣品的50μl/孔的心浸湯培養基中以50μl/孔添加所述稀釋了的各菌液,在37℃下靜置培養18小時。
培養完成后,通過濁度目視判斷菌是否增殖,求得菌株的mic。將結果示于3。
<試驗例4-4:針對流感嗜血桿菌測定mic>
進行所述各化合物針對流感嗜血桿菌的最小抑菌濃度(mic)的測定。
將流感嗜血桿菌(haemophilusinfluenzae)t-196株、和流感嗜血桿菌(h.influenzae)ard476株在hi培養基(在mullerhinton培養基(bectondickinson公司制)中添加5%的fildes強化營養(fildesenrichment,bectondickinson公司制))中、在37℃、含5%碳酸氣的需氧條件下靜置培養一夜。
培養完成后,將培養液在hi培養基中懸浮,以各流感嗜血桿菌為2×106cfu/ml~9×106cfu/ml的方式稀釋。
各試驗樣品(所述各化合物、克拉霉素、氨芐青霉素)分別在hi培養基中以256mg/l制備。由此,進行兩倍階段稀釋,進行11階段稀釋到0.125mg/l為止。
分別在含有所述各濃度的試驗樣品的50μl/孔的hi培養基中以50μl/孔添加所述稀釋了的各菌液,在37℃、含5%碳酸氣的需氧條件下靜置培養18小時。
培養完成后,通過濁度目視判斷菌是否增殖,求得菌株的mic。將結果示于3。
[表3]
如表3所示,所述各化合物顯示出抗幽門螺桿菌活性。在這些之中,結構式(3)表示的化合物、結構式(11)表示的化合物、結構式(12)表示的化合物均以低濃度顯示出抗幽門螺桿菌活性。
另一方面,所述各化合物針對其它通常的感染病原菌為弱的抗菌活性。
本發明的任一個結構式(1)~(13)表示的化合物具有優異的抗癌作用或優異的抗幽門螺桿菌活性,是安全性高的化合物,因此,可以優選用作藥品組合物、抗癌劑、抗幽門螺桿菌劑等有效成分。
- 還沒有人留言評論。精彩留言會獲得點贊!